


Department of Dermatology, Xinhua Hospital, Shanghai Jiaotong University School of Medicine, 1665 Kongjiang Road, Shanghai 200092, China. *E-mail: liming01@xinhuamed.com.cn, yaozhirong@xinhuamed.com.cn
Accepted Apr 2, 2019; E-published Apr 2, 2019
Congenital poikiloderma is a dermatological condition characterized in the first few months of life by epidermal atrophy, telangiectasias, and variegated pigmentation (hypo- and hyperpigmentation). Poikiloderma often presents a diagnostic challenge in the first few months of life, with differential diagnoses such as Rothmund-Thomson syndrome (RTS), the eponymous Weary form of hereditary sclerosing poikiloderma, Clericuzio-type poikiloderma with neutropaenia, and Kindler syndrome (1–3).
A distinct autosomal dominant form of hereditary fibrosing poikiloderma (HFP) was first described in a South African family in 2006 (3). HFP with tendon con-tractures, myopathy, and pulmonary fibrosis (abbreviated POIKTMP) is caused by mutations in FAM111B (Homo sapiens family with sequence similarity 111, member B), encoding a trypsin-like cysteine/serine peptidase (4, 5). FAM111B has also been implicated in susceptibility to prostate cancer (6). The main clinicopathological features of POIKTMP comprise early-onset poikiloderma, especially on the face and sun-exposed areas, alopecia, hypohidrosis with heat intolerance, growth retardation, multiple muscle contractures, in particular triceps surae muscle contractures, progressive muscle weakness, progressive pulmonary fibrosis, exocrine pancreatic dysfunction, liver impairment, cataracts and haematological abnormalities (4, 7–9).
This study, which was approved by the ethics committees of Shanghai Jiaotong University School of Medicine and conducted in accordance with the principles of the Declaration of Helsinki, describes the clinical and genetic background of a POIKTMP patient with FAM111B missense mutation.
We describe here a new case of POIKTMP in a 5-month-old Chinese boy. He was born of non-consanguineous Chinese parents, and had no similar illness history or familial history of atopy. He was the product of a full-term, uncomplicated pregnancy and delivery, with a birth-weight of 3.5 kg, with no immediate perinatal abnormalities.
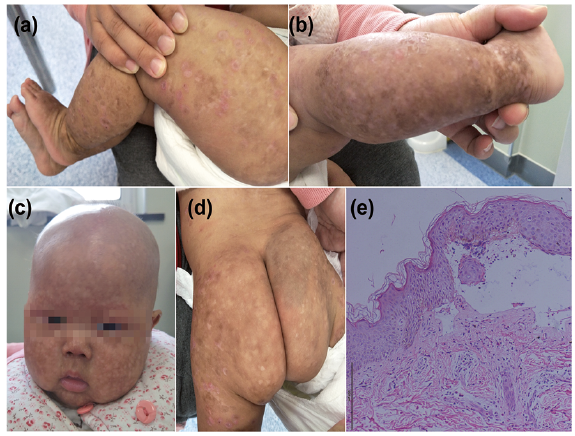
After 6 days, however, erythema, with desquamation on the cheeks with a recurrent papulovesicular facial eruption was noted. Thereafter the clinical features comprised progressive generalized poikiloderma, telangiectasia, xerosis and innumerable hypo- and hyperpigmentary macules, measuring between 3 and 6 mm, predominantly on the face and in other sun-exposed areas; and worsening non-scarring alopecia affecting the scalp, eyebrows and eyelashes (Fig. 1a–d). The patient also had eczematous lesions on the legs and feet. No lymphoedema of the upper and/or lower extremities was observed.
Fig. 1. Clinical features of hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis (POIKTMP) in a 5-month-old Chinese boy. (a–d) Aged 5 months, he exhibited generalized poikiloderma, and alopecia with a resolving papulovesicular eruption; eczema-like dermatosis was also present. (e) Haematoxylin and eosin (H&E)-stained sections of proband skin. Scale bar: 200 µm. Written permission to publish these photos are given.
A biopsy from the leg revealed conspicuous thickening of the spinous layer, blister formation under the epidermis, cellulose-like substance deposition and inflammatory cell infiltration in the blister with scattered eosinophils and neutrophils. In allergen detection, the patient was allergy to worm.
Other normal or negative findings included: weight and height within normal limit; full blood count; immunoglobulins; complement; anti-ds-DNA antibodies, anti-ANA antibodies, anti-CENPB antibodies; TPPA, RPR, anti-TP antibodies; no heat intolerance; no respiratory abnormalities; no liver impairment, no exocrine pancreatic insufficiency; no muscle weakness or wasting; no tendon abnormalities or joint contractures; no nail abnormalities; no cataracts; no recurrent gingivitis; no palmoplantar keratoderma.
Following informed consent, the genomic DNA of the proband was analysed using a gene probe consisting of 541 genetic loci of genodermatoses. This revealed a heterozygous missense mutation, c.A1873C in FAM111B, which results in the mutation p.Thr625Pro (Fig. 2a). No mutation was detected in his non-consanguineous parents (Fig. 2b, c). This mutation converts Thr625 to Pro and was not detected in his healthy parents or in 100 unrelated healthy Chinese individuals (200 alleles) by Sanger sequencing (10). The mutation was also absent from the public database (NCBI, dbSNP135, the 1000 Genomes Project, and HapMap8) and our internal datasets, suggesting that this was most likely the deleterious mutation in this patient. In summary, we identified a de novo missense mutation that is localized in the loop of the predicted functional domain of FAM111B.
Fig. 2. Molecular features. Sequencing revealed a de novo missense mutation, c.A1873C, in FAM111B, which was absent in his unaffected parents. (a) The proband, (b) his father and (c) his mother.
The phenotype of this case differs from other types of hereditary poikiloderma, such as RTS, WRN, Clericuzio-type poikiloderma with neutropaenia, and Kindler syndrome (2, 11–13). Neither did we identify potentially pathogenic mutations in specific genes, such as RECQL4 (mutated in Rothmund-Thomson syndrome), C16orf57 (mutated in Clericuzio-type poikiloderma with neutropaenia) or other genes implicated in inherited poikiloderma.
In 2013, Mercier et al. (4) identified the causative gene, FAM111B of POIKTMP. A previous individual (a French boy) was reported with the mutation c.1874C>A (p.Thr625Asn) and exhibited poikiloderma, hypotrichosis, alopecia and palmar erythrosis, since early childhood. No bullous lesions, eczema-like lesions, or ichthyosis-like lesions were observed, unlike in our case (8). The French boy also developed leg contractures at 7 years old, followed by leg weakness at 8 years old, and a pulmonary function test showed restricted syndrome.
With regard to genotype-phenotype correlation, codon 625 is located in the functional domain of FAM111B, the possibility being that mutation in the functional domain could be associated with earlier onset of the disease and a more severe phenotype in terms of cutaneous, muscle or visceral involvement (4, 8). Nevertheless, there were differences in our patient compared with the p.Thr625Asn case with regard to the limbs’ contractures, muscle weakness, and pulmonary restricted syndrome. However, many of the clinicopathological features of POIKTMP begin to manifest during childhood, and therefore the future clinical course of our patient is uncertain. Indeed, the clinical features of POIKTMP are somewhat variable (4).
In conclusion, we report here another case of POIKTMP, and expand our knowledge of this very rare entity. Further studies are needed to understand the function of FAM111B.
The authors would like to thank all patients who participated in this study.
This work was supported by a grant from the National Nature Science Foundation of China (81874239), grants from Shanghai Municipal Education Commission-Gaofeng Clinical Medcine Grant Support (20161417), grants from Medical Engineering Cross Research Foundation of Shanghai Jiaotong University (YG2017MS73), and Shanghai Sailing Program (16YF1407400).
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize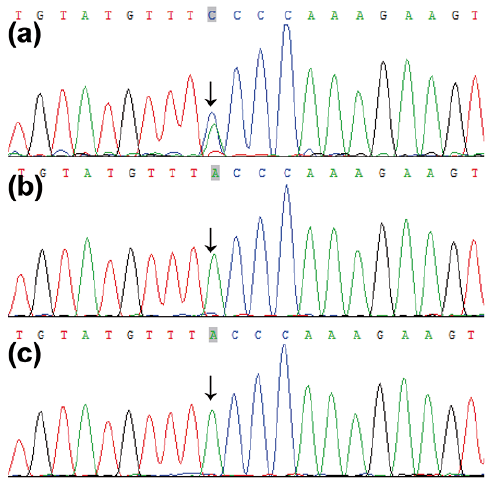 Click to show fullsize
Click to show fullsize