


1Department of Dermatology, Shiga University of Medical Science, Setatsukinowa, Otsu, Shiga 520-2192, and 2Division of Respirology, Neurology, and Rheumatology, Department of Medicine, Kurume University School of Medicine, Kurume, Fukuoka, Japan. *E-mail: noriki@belle.shiga-med.ac.jp
Accepted Apr 16, 2019; E-published Apr 16, 2019
Systemic lupus erythematosus (SLE) is a systemic autoimmune inflammatory disease characterized by antibody production against cellular nuclear elements (1). On the other hand, familial Mediterranean fever (FMF) is an auto-inflammatory disorder, triggered by FMF-associated point mutations. Recurrent high fever, generally above 38°C for several days, is the most important symptom of FMF accompanied by peritonitis, pleuritis, and arthritis without deformity. Although colchicine is effective, corticosteroids are not useful for symptom control (2). Fever, joint pain, plaques, and pleurisy are common manifestations of both of these diseases. Here, we present a rare case of SLE with FMF and a literature review.
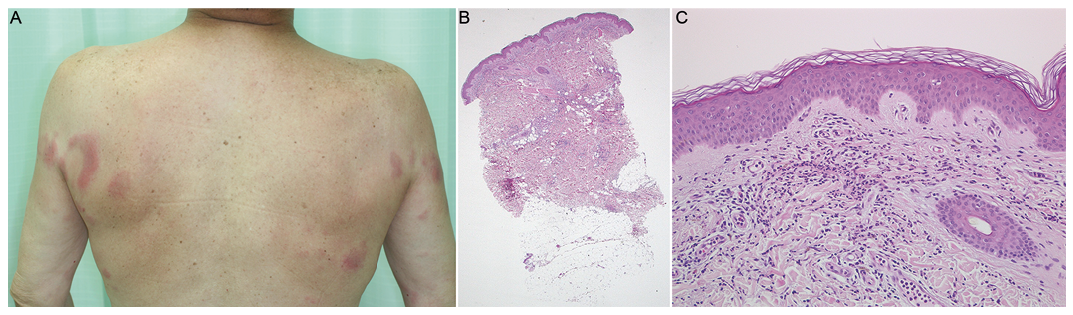
A 64-year-old Japanese man noticed joint pain in his shoulders, elbows, and knees. He has frequently presented with fever over 38°C and indurated erythema for several days about once a month since 2012. He has never experienced the same symptoms before, and his family demonstrated no similar symptoms. His local dermatologist diagnosed him with Sweet’s syndrome from clinical and histopathological features, and treated him with 5 to 20 mg a day of oral corticosteroids. However, due to the recurrence of symptoms he was referred to our hospital in 2013. Physical examination revealed persistent malar erythema and transient indurated erythema on the upper extremities and trunk (Fig. 1A). He had non-erosive arthritis on shoulders, elbows, and knees. He did not present with swollen fingers, alopecia, lymphadenopathy, or oral ulcers. On the first visit, the results of blood tests showed elevated levels of white blood cell counts (9,000/µl), serum C-reactive protein (2.46 mg/dl), IgG (1,910 mg/dl), and erythrocyte sedimentation rate (45.4 mm/h), but a decreased level of lymphocytes (927/µl). Serum levels of creatinine, creatine kinase and aldolase were within normal limits. Urine examination showed no abnormalities. Anti-nuclear antibody had a speckled pattern and the level was more than 2,560 times. Anti-U1 RNP antibody level was more than 300 U/ml. Autoantibodies against double stranded DNA, smith, SS-A, SS-B, and cyclic citrullinated peptide were not detected. A cutaneous biopsy specimen from the erythema on his buttock showed perivascular and lobular infiltration of lymphocytes and neutrophils in the dermis and subcutaneous tissue (Fig. 1B). Vacuolar changes in the basal layer and perifollicular inflammation were not observed (Fig. 1C). Direct immunofluorescence showed no deposits of complement and immunoglobulin. Computed tomography revealed mild interstitial pneumonia, which we treated with low dose oral corticosteroids. We initially diagnosed this case as SLE because of malar erythema, non-erosive arthritis, lymphopenia and positive antinuclear antibody, based on the criteria (1), and denied mixed connective tissue disease due to the absence of swollen fingers and myositis. We added tacrolimus, since antimalarial drugs were not available in Japan. After a short while, he complained of precordial pains, due to pleurisy. We performed a mutational analysis of the familial Mediterranean fever gene (MEFV), which showed heterozygous mutations in exon 3; p.Arg408Gln and p.Pro369Ser. We suspected an atypical FMF and additionally administered 0.5 mg dose of colchicine. His precordial pains and fever improved even after the discontinuation of tacrolimus, and we were able to reduce the dosage of corticosteroids. We finally diagnosed the case as SLE with atypical FMF. When the dosage of colchicine increased, he got moderate liver damage. We continued with 0.5 mg dose of colchicine. We could not increase the dosage of colchicine above 10 mg a day due to liver dysfunction. Corticosteroids could not be decreased to less than 7 mg a day because of interstitial pneumonia. Thereafter, he has not complained of repeated fever and precordial pains.
Fig. 1. (A). Clinical presentation on the first visit. Indurated erythemas were observed on upper extremities and trunk. (B). Histopathological examination showed superficial and perivascular infiltration of lympho-cytes throughout the dermis and subcutaneous tissue (hema-toxylin-eosin (H-E) staining, x40). (C) Vacuolar changes in the basal layer and perifollicular inflammation were not observed (H-E x200).
We considered this case as a combination of SLE and FMF for several reasons. Firstly, there are no reports of SLE patients having the MEFV gene mutations on exon 3 contrary to exon 10 or exon 2 (3). Secondly, his symptoms improved with colchicine. Third reason is that the positivity of anti-U1 RNP antibody does not occur in FMF patients. Lastly, interstitial pneumonia does not likely occur in FMF patients.
Differential diagnoses of the indurated erythema include cutaneous lupus erythematosus, Sweet’s syndrome, and neutrophil dermatosis. The typical skin lesion of FMF is a well-defined erythematous plaque like erysipelas. Sweet’s syndrome-like eruption has also been reported. Common histological features include slight edema of the superficial dermis and sparse perivascular infiltrate composed of lymphocytes, neutrophils, and histiocytes (4). In our case, the eruptions transiently appeared for several days along with fever. Histopathological examination showed perivascular and lobular infiltration of lymphocytes and neutrophils in the dermis and subcutaneous tissue without vacuolar changes in the basal layer and perifollicular inflammation, and lupus band test was negative. Therefore, we diagnosed the indurated erythema on the upper extremities and trunk as skin manifestations of FMF.
The MEFV gene variations are reported to be found also in patients with SLE, which revealed that the 4 MEFV (M694V, M680I, V726A and E148Q) gene variations are observed in 15% of patients with SLE and 10% of healthy controls (3). There are some papers speculating that the high levels of CRP in FMF patients act on removing the apoptotic materials, which may prevent the development of SLE.
To our knowledge, there have been only 21 published cases, including our case, on the coexistence of FMF and SLE as shown in Table SI (5–18). Only 2 cases, including ours, are male patients, probably due to the low male-to-female ratio in SLE. Although abdominal pain is noted in 16 cases, only our case has chest pain. It is stated that atypical FMF needs a larger dose of colchicine than the typical one; however, comparatively lower doses of colchicine are being prescribed for SLE with FMF patients. While 31.3% of Japanese FMF patients had mutations in other than exon 10 (2), all 4 Japanese SLE cases with FMF had mutations in exon 2 or 3. Although further investigations are needed, the mutations of MEFV in exon 2 or 3 might be associated with the development of FMF in Asian SLE patients. The types of gene mutations may be related to the characteristics such as onset (4), symptoms, and complications. It is expected that a future study will reveal the mechanism of FMF through these case reports and review of literatures.
As a result, the mutational analysis of MEFV led to the successful treatment with colchicine in our case. We should watch out for FMF when we encounter SLE patients who do not respond to standard treatments or suffer from chest pain with recurrent high fever for a few days. We must be careful not to misdiagnose FMF in refractory SLE patients like our case.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize