


1New Children’s Hospital, University of Helsinki and Helsinki University Hospital, 2Rare Disease Center, New Children’s Hospital, University of Helsinki and Helsinki University Hospital, 3Blueprint Genetics, 4Folkhälsan Research Center, Helsinki, Finland, 5Immunobiology of Dendritic Cells, Inserm U1223, Institut Pasteur, Paris, France, 6Department of Pathology, Cancer and Translational Medicine Research Unit, University of Oulu, 7Department of Pathology, Oulu University Hospital, Oulu, 8Stem Cells and Metabolism Research Program, University of Helsinki and Folkhälsan Research Center, 9Department of Radiology, Helsinki Medical Imaging Center, Helsinki University Hospital, and 10Department of Dermatology and Allergology, University of Helsinki and Helsinki University Hospital, Helsinki, Finland
#These authors contributed equally and should be considered as first authors.
Desmoplakin (DSP) and Desmoglein 1 (DSG1) variants result in skin barrier defects leading to erythroderma, palmoplantar keratoderma and variable other features. Some DSG1 variant carriers present with SAM syndrome (Severe dermatitis, multiple Allergies, Metabolic wasting) and a SAM-like phenotype has been reported in 4 subjects with different heterozygous DSP variants. We report here a patient with a novel DSP spectrin region (SR) 6 variant c.1756C>T, p.(His586Tyr), novel features of brain lesions and severe recurrent mucocutaneous herpes simplex virus infections, with a favourable response to ustekinumab. Through a review of reported cases of heterozygous variants in DSP SR6 (n = 15) and homozygous or compound heterozygous variants in DSG1 (n = 12) and SAM-like phenotype, we highlight phenotypic variability. Woolly hair, nail abnormalities and cardiomyopathy characterize patients with DSP variants, while elevated immunoglobulin E and food allergies are frequent in patients with DSG1 variants. Clinicians should be aware of the diverse manifestations of desmosomopathies.
Key words: desmoglein; desmoplakin; metabolic wasting; SAM syndrome; severe dermatitis.
Accepted Apr 29, 2019; E-published Apr 29, 2019
Acta Derm Venereol 2019; XX: XX–XX.
Corr: Svetlana Vakkilainen, New Children’s Hospital, University of Helsinki and Helsinki University Hospital, Stenbäckinkatu 9, FIN-00290 Helsinki, Finland. E-mail: svetlana.vakkilainen@hus.fi
Desmoplakin (DSP) and Desmoglein 1 (DSG1) gene changes result in skin barrier defects leading to widespread red scaly rash, skin thickening on the palms and soles and variable other features. We report here a patient with a novel DSP gene change, novel features of brain lesions and severe viral infections, and a favourable response to treatment with ustekinumab. Woolly hair, nail abnormalities and heart problems characterize patients with DSP gene changes, while elevated serum IgE levels and food allergies are frequent in patients with DSG1 gene changes. Clinicians should be aware of the diverse consequences of DSP and DSG1 gene abnormalities.
Desmoplakin and desmogleins form the integral parts of desmosomes, which are adhesive intercellular junctions crucial in tissues prone to mechanical stress (e.g. skin, heart, gastrointestinal mucosa) (1). Desmosomal proteins play a role in cell signalling and skin barrier function.
Desmoglein 1 (DSG1) and Desmoplakin (DSP) encode critical components of desmosomes, and pathogenic variants in both genes have been implicated in inflammatory skin disorders. DSG1 variants have been reported recently in patients with designated SAM syndrome (Severe dermatitis, multiple Allergies and Metabolic wasting) with an extremely variable phenotype consisting of severe erythrodermic dermatitis, failure to thrive (FTT), recurrent infections, metabolic wasting, multiple allergies, increased immunoglobulin (Ig) E levels and eosinophilia (2–7).
Two patients with different heterozygous DSP variants were reported to have a SAM-like phenotype (8, 9). Autosomal dominant DSP variants within the same spectrin 6 (SR6) region also cause erythrokeratoderma and cardiomyopathy (10), and palmoplantar keratoderma (PPK) with woolly hair, cardiomyopathy and arrhythmias (11). Recently, 2 additional patients with heterozygous DSP SR6 variants have been reported to display SAM syndrome, together with ectodermal dysplasia and cardiomyopathy (coined SAMEC syndrome) (12).
The lack of epithelial barrier proteins commonly seems to lead to immunological dysregulation. Impaired epithelial barrier function enhances Th2 responses and leads to chronic activation of the immune system (13). DSG1 deficiency results in increased expression of various genes encoding cytokines involved in allergic manifestations, such as IL5 and TNF (2). In SAMEC, DSG1 was linked to an inability to retain ERBB2-interacting protein (ERBIN) at the cell membrane, probably hampering NF-κB pathway inhibition and promoting epithelial inflammation (12). Heterozygous DSP variants associate with abnormally high levels of proinflammatory cytokines (interleukin (IL) 6, IL-8, and IL-1β), NF-kB target gene products, and thymic stromal lymphoprotein in the keratinocytes (12). The dysregulated epidermal immune environment triggers early-onset allergic manifestations beyond the skin, including asthma and allergic rhinoconjunctivitis, as well as eosinophilic oesophagitis and colitis similar to patients with Netherton syndrome (14).
Desmoplakin anchors intermediate filaments to desmosomal plaques and is expressed at high levels in the skin and heart muscle, and at medium levels in bronchi and oesophagus (15). Importantly, individuals with DSP and other desmosomal gene variants are at risk of developing cardiomyopathy and lethal arrhythmias, typically at the age of 3–12 years (16), warranting continuous cardiac follow-up.
We report here a patient with a novel DSP SR6 region variant (c.1756C>T, p.His586Tyr), presenting with erythro-dermic ichthyosis, severe FTT, oesophagitis, recurrent infections, and novel features, such as severe recurrent mucocutaneous herpes simplex virus (HSV) infections, pyogenic granuloma on the tongue, gastritis and brain lesions, further diversifying the DSP-related phenotype. We also reviewed all reported patients with heterozygous variants in DSP SR6 and with homozygous or compound heterozygous variants in DSG1 and SAM/SAMEC-like phenotype, in order to delineate variable and overlapping clinical features.
Written informed consent was obtained from the patient’s parents, including permission to publish the photographs.
The immunological assessment, skin histopathology and immunohistochemistry, hair analysis, oesophagus and gastric biopsies, whole-exome sequencing and variant analysis, protein modelling, as well as literature search and statistical analyses, are described in detail in Appendix S1.
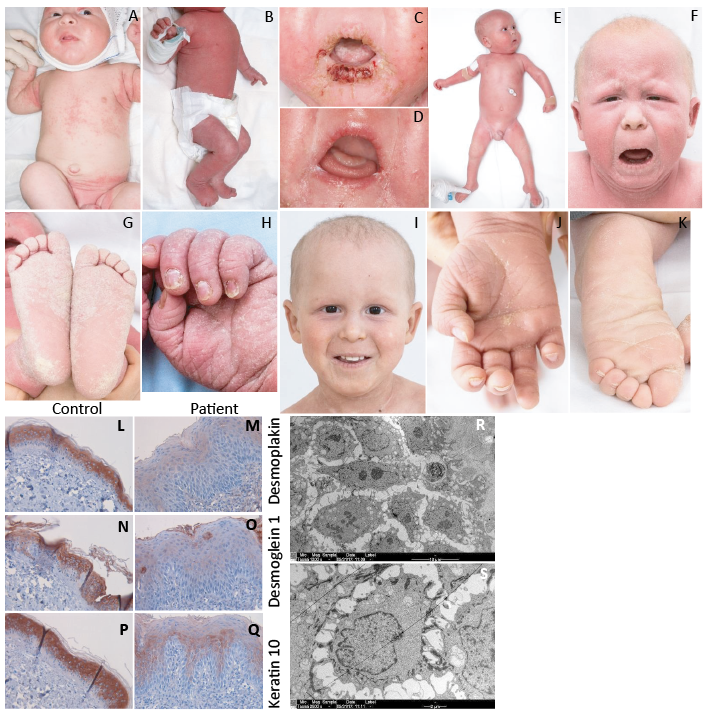
Our patient is the first child of non-consanguineous parents of Arab descent, born at term with normal weight and length. At 2 weeks of age, he developed an intertriginous seborrhoeic eczema-like dermatitis (Fig. 1A). A pustular eruption emerged at age 1.5 months and rapidly evolved into a severely pruritic and seemingly painful ichthyosiform erythroderma (Fig. 1B). His hair was normal at birth, but total alopecia developed at 3 months. His nails were abnormally thin and soft.
He had had relapsing Staphylococcus aureus skin infections, Candida albicans thrush, recurrent enterococcal and staphylococcal sepsis since the age of 4 months. At 5 months, he developed severe and relapsing protracted HSV-1 stomatitis with extensive mucocutaneous involvement (Fig. 1C). Extensive infectious, immunological and metabolic evaluations were mostly normal and did not suggest any specific immunodeficiency syndrome.
From age 4 months onwards, he developed difficulties with swallowing and progressive FTT. These necessitated percutaneous gastrostomy and initiation of amino acid-based formula feeding. Cow milk protein intolerance was diagnosed at 7 months by an in-hospital oral provocation challenge and manifested as profuse vomiting, diarrhoea and inconsolable crying within hours of administration of standard formula. At 6 months, his serum IgE levels increased up to 958 kU/l (normal < 70 kU/l), but then gradually returned to near normal value (102 kU/l). Eosinophil counts remained normal. In the ImmunoCAP specific IgE assay against 112 allergens, only a low-level reactivity (0.7 ISU, normal > 0.3 ISU) to prohevein rHev b 6.01 was detected at 9 months, but at 11 months positivity to peanut (0.37 kU/l) and egg-white (1.03 kU/l) appeared. Profound fluid balance disturbances with hypo- and hypernatremia and rapid dehydration or generalized oedema occurred during erythrodermic flares. Arrhythmia or cardiac abnormalities have, so far, not been detected.
Upper gastrointestinal tract endoscopy was normal at 4 months, but revealed lymphocyte and plasmablast mucosal infiltrates in the stomach at 7 months. After several courses of proton pump inhibitors, his gastric histology normalized at 2 years of age. However, he then exhibited mild oesophagitis with some intraepithelial eosinophils, not numerous enough to fulfil the criteria for eosinophilic oesophagitis.
Motor development was delayed from the age of 4 months. Brain magnetic resonance imaging (MRI) at 4 months showed symmetrical diffusion restriction in the superior cerebellar peduncles and decussation. Also, red nuclei in mesencephalon, subthalamic region, globi pallidi and optic radiation were involved (Fig. S1). Repeated MRI a month later showed no progression of these changes. His motor development progresses, but remains delayed, with no independent walking at 2 years of age.
At 6 months he developed a firm papular lesion 2.0 cm in diameter on his tongue (Fig. 1D), which resolved spontaneously over months. Biopsy of the lesion revealed a pyogenic granuloma. HSV-1, cytomegalovirus and human papillomavirus (HPV) 11 were found in the lesion by nucleic acid assay, while other viral, bacterial, mycobacterial and fungal investigations were negative.
The patient’s skin was treated with basic emollients and courses of topical mild potency corticosteroids. McAleer et al. (8) reported topical tacrolimus, systemic antibiotics, intravenous immunoglobulin (IVIG) therapy and systemic acitretin to be effective in their patient with a DSP variant. At 5 months, IVIG therapy for recurrent sepsis was commenced. He has had no bacterial infections since, while recurrent mucocutaneous HSV-1 flares have necessitated continuous acyclovir prophylaxis. IVIG substitution was stopped at 20 months. Acitretin was started at a low dose of 0.25 mg/kg every other day at 7.5 months (8). Acitretin slightly alleviated scaling, erythema and itch and induced hair regrowth (Fig. 1E). Due to constant vomiting, acitretin was stopped at 14 months, followed by no significant change in his skin condition nor vomiting. His first tooth erupted at 10 months and his teeth have been normal. At 18 months, he developed ichthyotic erythroderma (Fig. 1F–H), and, additionally, he has developed mild PPK and onychodystrophy. His growth has improved substantially due to optimized enteral nutrition.
Fig. 1. Clinical features of the reported patient. (A) An intertriginous seborrhoeic like eczema at 2 weeks of age (B) evolved into a ichthyosiform erythroderma at 1.5 months. (C) Severe and relapsing HSV-1 stomatitis at 5 months and (D) a pyogenic granuloma on the tongue at 6 months. (E) Slight alleviation of scaling and erythema and hair regrowth after initiation of acitretin therapy at 7 months. (F) Ichthyotic erythroderma at 18 months with the development of (G) slight palmoplantar keratoderma (PPK) and (H) onychodystrophy. (I–K) Response to ustekinumab treatment at 25 months included hair regrowth and alleviation of skin scaling and PPK. DSP mutation leads to decreased levels of DSP, DSG1 and KRT10. (L) Skin tissue of a normal control shows prominent DSP antibody staining throughout the epidermis in suprabasal cells, while in the patient (M) only a weak staining is seen in the upper epidermis. DSG1 is distinct intercellularly in (N) healthy skin, but almost totally lost (O) in the patient. KRT10 stains intensely in (P) the suprabasal healthy skin, but is weak in the patient’s upper epidermis and (Q) diminished in the suprabasal layers. Electron microscopy of the patient’s skin biopsy samples shows the loss of intercellular cohesiveness, especially (R) suprabasally and decreased numbers of desmosomes and tonofilaments, as well as (S) the absence of perinuclear tonofilaments. Written permission from the parent.
Since 2 patients with DSP variants were reported to benefit from ustekinumab therapy, with reduction in itching and in dermatitis severity and improvement in hair thickness (9), we initiated ustekinumab treatment at 22 months in our patient. Ustekinumab (0.75 mg/kg) was given initially at 4 weeks and from then on every 12 weeks, leading to alleviation of pruritus, skin scaling and PPK, as well as better hair growth after the first 2 injections (Fig. 1I–K). Oral intake also improved significantly, and gastrostomy use could be discontinued. Along with clinical improvement, serum levels of IL-17A, IL-17F and IL-23 decreased strikingly, whereas concentrations of IL-12p70 somewhat increased, albeit at very low levels (< 1 pg/ml) (Fig. S2).
Skin biopsy findings. Skin biopsy from the pustular eruption at 1.5 months showed eczema with a neutrophilic infiltrate and microabscesses. Biopsy at 3 months from the erythrodermic skin revealed a psoriasiform reaction pattern with a dense ichthyosiform stratum corneum and neutrophilic infiltrates in the epidermis and upper dermis. Filaggrin immunostaining was reduced in the first and absent in the second biopsy, suggesting ichthyosis, while LEKTI immunostaining was normal in both biopsies (data not shown). Hair morphology was normal. Immunohistochemistry demonstrated significant decrease in desmoplakin, desmoglein 1 and keratin 10 (Fig. 1L–Q). Skin electron microscopy revealed decreased numbers of desmosomes and tonofilaments, and perinuclear tonofilaments were absent. The loss of intercellular cohesiveness was especially prominent suprabasally (Fig. 1R–S).
Genetic work-up. Ichthyosis and primary immunodeficiency gene panels (Blueprint Genetics, helsinki, Finland) detected no pathogenic variants. Clinical whole-exome sequencing was performed with median sequencing coverage of 132× and 99.1% of target regions were covered with at least 20 reads. Analysis of whole-exome sequence variants revealed a novel heterozygous missense variant in the DSP gene: c.1756C>T, p.(His586Tyr) (NM_004415.2), confirmed with bidirectional Sanger sequencing. Based on Sanger sequencing analysis, the parents were not carriers, thus the variant was de novo in the patient. This variant has not been reported in the Exome Aggregation Consortium (ExAC) database or Genome Aggregation Database (gnomAD). The variant is predicted deleterious by in silico tools SIFT and MutationTaster and benign by PolyPhen, the CADD score (PHRED-like scaled C-score) is 24.3, suggesting that the amino acid change is deleterious. The His586 residue is located within one of the spectrin repeat domains, SR6, which is a hotspot for dominant missense variants causing cutaneous disease (10). Historical patient with a similar phenotype carried a de novo missense variant in the same amino acid residue p.(His586Pro) (8). Protein modelling with the template-based protein structure tools RaptorX (17) and Swiss-model (18) did not predict alterations in the protein structure. However, histidine is an amino acid with unique chemical properties not particularly well substituted with any other amino acid.
Clinical whole-exome sequencing also identified a variant of uncertain significance, a novel heterozygous in-frame deletion c.11361_11387del, p.(Asp3788_Thr3796del) (NM_001376.4) in DYNC1H1, which encodes the heavy chain of cytoplasmic dynein 1 that acts a motor involved in trafficking of vesicles and organelles along microtubule. The variant deletes 9 highly conserved amino acid residues in the motor domain of DYNC1H1 (19). Sanger sequencing of both parents revealed that the variant has occurred de novo in the patient.
The clinical and laboratory characteristics of the patients were compared with heterozygous variants in DSP SR6 and with homozygous or compound heterozygous variants in DSG1 and SAM-like phenotype (Table I and Fig. 2). SR6 region dominant DSP variants have been reported in 15 patients, with clinical features ranging from a constellation of PPK, woolly hair and arrhythmogenic cardiomyopathy at one end of the spectrum (patients 7–13) to a SAM-like disease with erythroderma, ichthyosis, hypotrichosis, recurrent infections, FTT and gastrointestinal manifestations (patients 1–3, 14) at the other end of the spectrum (Table I). Overlapping features of cardiomyopathy, erythroderma, ichthyosis, hypotrichosis, PPK and woolly hair have been reported in another 4 patients with DSP variants (patients 4–6, 15, Table I).
Table I. Clinical and laboratory characteristics of patients with heterozygous variants in DSP SR6
Fig. 2. The prevalence of clinical features in 15 patients with DSP (black bars, Table I) and 12 patients with DSG1 (grey bars, references 2–7) variants.
Overall, the shared clinical manifestations (DSP/DSG1) are as follows: PPK (100%/92%), erythroderma (53%/67%), ichthyosis (53%/50%), FTT (40%/33%), recurrent infections (20%/33%), gastrointestinal problems (27%/17%) and developmental delay (20%/25%) (Fig. 2). Otherwise, clinical features varied considerably. Subjects with DSG1 variants do not manifest eosinophilia, pustulosis, dental and ophthalmic abnormalities, hoarse voice, cardiomyopathy or arrhythmias. Woolly hair and nail abnormalities were more common in patients with DSP variants (14/15 vs. 4/12, p = 0.003 and 11/15 vs. 1/12, p = 0.001, respectively), while elevated IgE and food allergies were observed more frequently in patients with DSG1 variants (10/12 vs. 3/15, p = 0.002 and 8/12 vs. 3/15, p = 0.022, respectively).
We report here a patient with a novel DSP variant c.1756C>T, p.His586Tyr and severe erythrodermic ichthyosis, resembling, but distinct from, SAM, with previously undescribed features of severe recurrent mucocutaneous HSV-1 infection, oral pyogenic granuloma, gastritis and brain lesions. The patient was treated successfully with ustekinumab, targeting the IL-12 and IL-23 as well as downstream IL-17 pathways and adding to the previously reported favourable experience with this medication in another 2 patients with DSP SR6 variants (9). The current patient is thus the third case reported to benefit from ustekinumab therapy.
Patients with SAM present with chronic inflammation of the skin and gastrointestinal epithelium, leading to a dysregulated immune response (9, 12). Our patient showed lymphocytic and plasmablast infiltrates in the gastric mucosa. Coupled with swallowing difficulties and gradually progressing vomiting, as well as further development of mild oesophagitis, these may reflect desmoplakin-related disruption of gastrointestinal mucosa or food intolerance. The severe reaction to cow’s milk was out of proportion to his serum IgE levels and resembled food protein-induced enterocolitis syndrome (23).
The aetiology of brain abnormalities in our patient remains unexplained, since DSP is not expressed in the central nervous system (The Human Protein Atlas; GTEx Portal) and since DSP variants have not been associated with brain tissue pathology. Therefore, factors such as undiagnosed perinatal hypoxia, severe FTT and drastic dysnatraemia may contribute. However, developmental delay has been reported in association with some DSP variants (Table I) and its link to desmosomal protein dysfunction warrants further study. Interestingly, our patient had a novel heterozygous de novo inframe deletion p.(Asp3788_Thr3796del) in DYNC1H1, of which de novo variants have been reported in patients with malformations of cortical development and various other neurological disorders (24, 25). However, the contribution of the DYNC1H1 variant to aetiology of the observed symmetrical bilateral diffusion modality changes in our patient remains unknown.
The self-resolving tongue pyogenic granuloma in our patient was positive for multiple viral nucleic acids. Although HPV is a well-known cause of mouth papillomatous lesions, both HPV and CMV nucleic acids are frequently detected in oral cavities of healthy infants (26, 27). Instead, HSV-2 and HSV-1 have been reported to cause self-resolving pseudotumour on the tongue (28) and pyogenic granuloma (29), consistent with the preceding HSV-1 mucocutaneous infection in our patient. Also, mechanical stress from a recently introduced pacifier may have contributed to lesion formation (30).
IVIG therapy may have reduced the number of bacterial infections in our patient, as well as in the first described individual with SAM due to DSP variant, despite the normal pretreatment IgG levels in both. Also, IVIG has demonstrated efficacy in patients with Netherton syndrome (31). This may reflect the need for higher IgG levels in patients with epidermal barrier defects who lose protein continuously via inflamed skin. In addition, specific antibody deficiency has been described in some subjects with Netherton syndrome (32). In our patient, IgG levels decreased despite initiation of IVIG, but stabilized after higher and more frequent IVIG dosing. The beneficial effects of IVIG therapy in our case may thus be explained by age-specific immature antibody production exacerbated by chronic protein loss via skin. Acitretin is successfully used in the management of ichthyoses (33, 34) and was also partially effective in our patient.
Ustekinumab targets the disrupted balance of IL-23 /Th17 pathway and has been efficient in controlling skin inflammation in 2 patients with DSP SR6 variants (9), as well as in our case. We provide novel data on the decrease in serum IL-17A and IL-17F levels after initiation of ustekinumab therapy, adding evidence of its biologic efficacy in patients with DSP mutations. The increase in IL-12p70 levels, although in levels below 1 pg/ml, during ustekinumab therapy may reflect a compensatory protective effect of IL-12 in skin inflammation (35). Follow-up studies are needed to test the efficacy, if any, of ustekinumab in preventing the development of cardiomyopathy, a fatal complication seen in the majority of DSP SR6 variant patients (Table I).
Patients with SR6 DSP variants manifest a spectrum of clinical features, ranging from PPK and cardiomyopathy to SAM-like disease. We add severe HSV-1 infection, brain abnormalities and persistent gastrointestinal problems to the diversity of their phenotype. DSP variants should be included in the differential diagnosis of infants with ichthyosiform erythroderma, FTT, recurrent infections and these novel features, to allow rapid diagnosis and early therapy. Based on current literature, the clinical features in SAM, SAMEC and SAM-like phenotypes are highly variable. Clinicians caring for these patients should be familiar with the diverse manifestations of desmosomopathies, as well as the importance of early genetic diagnosis, given that only patients with DSP SR6, not DSG1 variants, are at inevitable risk of developing cardiomyopathy. Further studies on the mechanisms of immune dysregulation in patients with DSG1 and DSP variants would provide background for novel targeted therapies, such as immunosuppressants or immunomodulators.
The authors thank Dr Isabelle Meyts for her critical comments concerning this case. DD acknowledges ImmunoQure for provision of L17F monoclonal antibodies.
The study was funded by the Doctoral School in Health Sciences at the University of Helsinki (SV), and Helsinki and Uusimaa joint authority research grant (TYH 2015210, AR, KHJ).
Conflicts of interest: MM is employed by Blueprint Genetics.


 Click to show fullsize
Click to show fullsize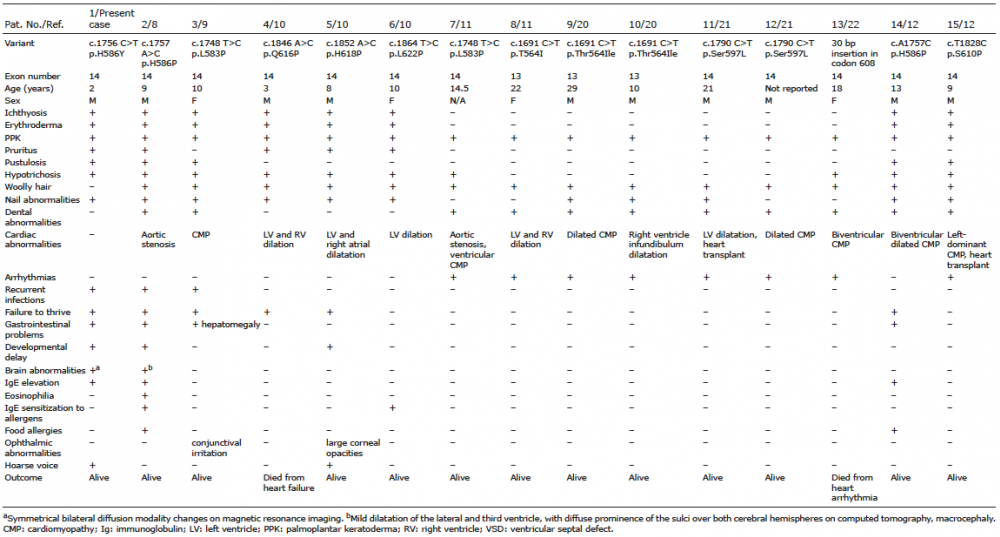 Click to show fullsize
Click to show fullsize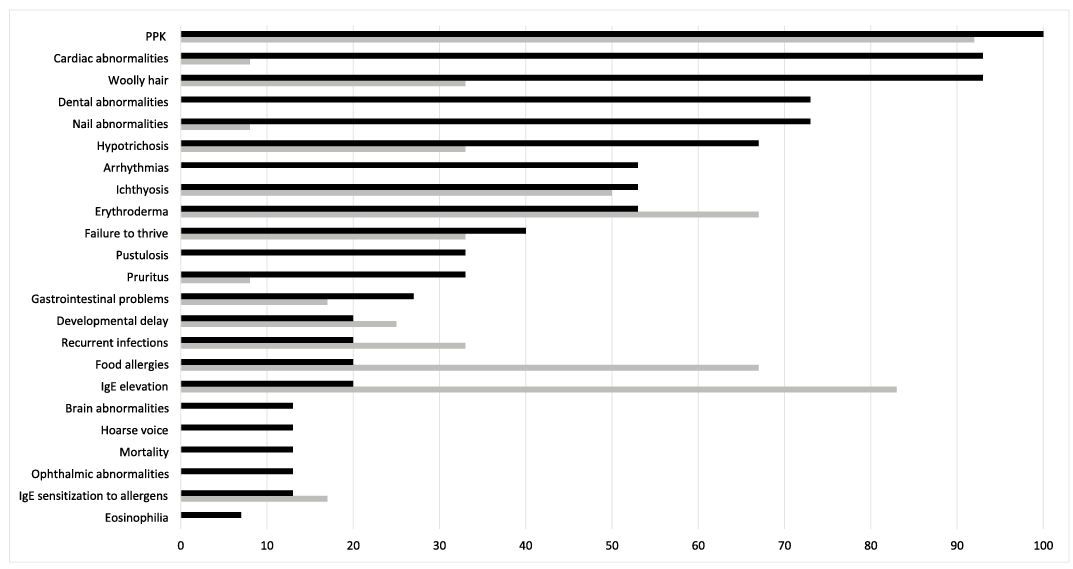 Click to show fullsize
Click to show fullsize