


1Laboratory of Molecular and Cell Biology, IDI-IRCCS, via dei Monti di Creta 104, IT-00167 Rome, 2Medical Genetics Unit and Laboratory of Medical Genetics, 3Pediatric Cardiology and Cardiac Arrhythmia/Syncope Unit, Department of Pediatric Cardiology and Cardiac Surgery, 4Dermatology Unit, and 5Genetics and Rare Diseases Research Division, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy. E-mail: l.guerra@idi.it
#These authors equally contributed to the work
Accepted May 9, 2019; E-published May 10, 2019
The association of woolly hair and palmoplantar keratoderma (PPK) has been found in 2 autosomal recessive syndromic disorders: Naxos diseases (OMIM 601214) and Carvajal syndrome (OMIM 605676), both also presenting progressive arrhythmogenic cardiomyopathy. Naxos disease is characterized by the triad of diffuse PPK, woolly hair and right ventricular (RV) arrhythmogenic cardiomyopathy (AC) and is caused by homozygous mutations in the plakoglobin gene (JUP) (1). Carvajal syndrome manifests with striated and fokal PPK, woolly hair and left-dominant dilated cardiomyopathy and is due to recessive mutations in the desmoplakin gene (DSP) (2). Few patients with these syndromes have been described (3–6). AC due to autosomal dominant DSP mutations, with (OMIM: 615821) or without (OMIM 607450) hypo/oligodontia, may be also associated with PPK and woolly hair (5–7).
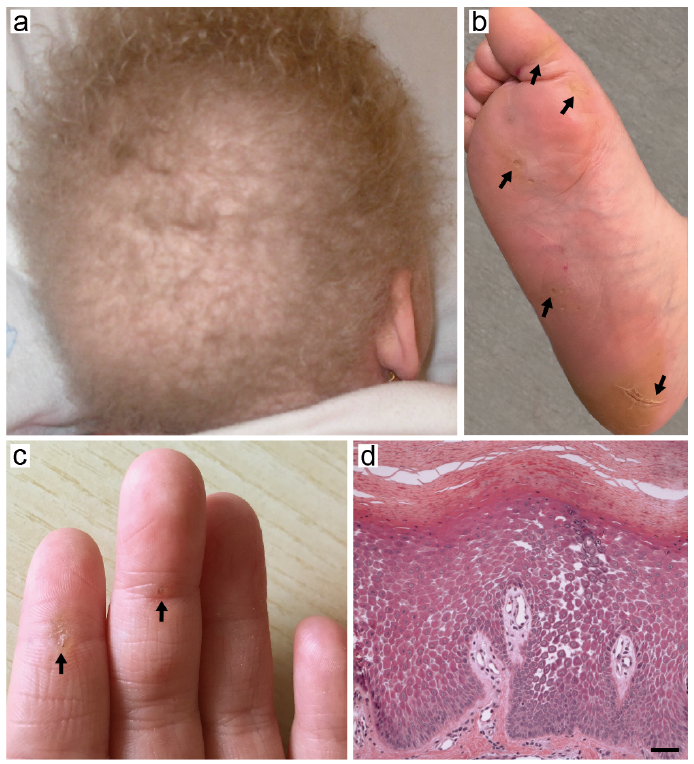
A 5-year-old female child first presented for hypotrichosis to the Rare Skin Disease outpatient clinic of IDI-IRCCS. She was born at term from healthy non-consanguineous parents. Physical examination revealed light-coloured woolly, sparse and fragile hair, which had never required cutting (Fig. 1a), focal plantar hyperkeratosis localized at pressure areas (Fig. 1b) and minimal striated hyperkeratosis of the palmar aspect of the first 3 digits of the hand (Fig. 1c). The patient also had dry skin and mild keratosis pilaris of the arms and thighs. Her parents reported that hair abnormalities had been present since birth, while plantar keratoderma manifested approximately at 1 year of age, followed by palmar lesions. Her nails and teeth were normal. The patient was otherwise in good general health. Histological examination of a plantar hyperkeratosis biopsy revealed massive hyperkeratosis, parakeratosis and acanthosis of the epidermis with acantholytic clefts between neighbouring keratinocytes and widened intercellular spaces in the suprabasal cell layers (Fig. 1d). Although histopathological findings were highly suggestive for involvement of a desmosomal protein component, molecular testing for the corresponding genes was not available at that time in Italy. The patient was therefore discharged with a diagnosis of epidermolytic PPK with woolly hair, and the recommendation to have a cardiological evaluation and follow-up in Sicily where the family lived. An electrocardiogram (ECG) and echocardiogram did not reveal any abnormality. Five years later (age 10 years), the family was contacted again as in the meantime molecular diagnosis became available at Bambino Gesù Children’s Hospital. Following written informed consent, genomic DNA was extracted from EDTA-blood and submitted to next-generation sequencing of a gene panel associated with cardiomyopathies (DES, DSC2, DGS2, DSP, JUP, LAMP2, PKP2). The analysis revealed 2 heterozygous truncating mutations in DSP (NM_004415): c.4788delA (p.Glu1597Serfs*5) and c.6091_6092delTT (p.Leu2031Glyfs*29) affecting exon 23 and 24, respectively. The former mutation was inherited from the father and the latter from the mother, as confirmed by Sanger sequencing (Fig. S1). The patient was then referred to the Cardiology and Arrhythmology Unit. Cardiological examination findings are presented in Table SI and Fig. S2. After a complete cardiac work-up, Carvajal syndrome was diagnosed on the basis of combined phenotype of woolly hair, striate PPK, left-dominant arrhythmogenic cardiomyopathy and molecular analysis findings. Therefore, the patient received an implantable subcutaneous cardioverter-defibrillator (ICD) and started anti-congestive heart failure treatment with angiotensin-converting enzyme inhibitors, beta-blockers and diuretics. At follow-up visits, the patient was in a fair general condition.
Fig. 1. Clinical and histopathological findings. (a) Woolly and sparse hair. (b, arrows) Focal plantar hyperkeratosis localized at pressure areas. (c, arrows) Minimal striated hyperkeratosis of the palmar aspect of the first 3 digits of the hand. (d) Hyperkeratosis with focal parakeratosis and suprabasal epidermal acantholysis in the haematoxylin-eosin staining. Bar: 50 μm.
We describe here a patient presenting the triad of woolly hair, PPK and biventricular arrhythmogenic cardiomyopathy due to compound heterozygous DSP mutations. The c.6091_6092delTT deletion (p.Leu2031Glyfs*29) is a previously reported DSP mutation described in compound heterozygosity with c.6079C>T (p.Arg1934*) mutation in a lethal acantholytic epidermolysis bullosa case (8). It results in truncation of the C-terminal tail of all 3 desmoplakin isoforms, I, Ia and II (9). The novel c.4788delA falls within the rod domain of desmoplakin and predicts truncation of only isoform I, while it spares the isoforms Ia and II that lack amino acid regions 1351-1793 and 1195-1793, respectively. Its functional consequences at protein level are expected to be similar to c.4778_4790del (p.Lys1593Serfs*5) mutation identified in compound heterozygosity with c.6310delA deletion in a patient presenting with skin fragility, PPK, alopecia and cardiomyopathy (10). Genotype-phenotype correlations in disorders with mutant DSP cannot be easily predicted. Truncating mutations in most patients developing cardiac disease are located in exons 23 and 24 (5, 6). In our patient both mutations are predicted to truncate the 3 C-terminal plakin-repeat subdomains from the longest desmoplakin (DSPI) isoform, resulting in protein loss-of-function. Indeed, plakin repeats are essential for coalignment and binding of intermediate filaments (11). Ablation of only DSPI, the major isoform expressed in the heart, due to the homozygous exon 23 mutation p.Arg1267* was shown to result in a Naxos-like syndrome associated with a severe heart phenotype in a child (12). Thus, it seems that DSPII can only in part compensate the function of the full isoform in both cardiac tissue, where it is almost absent, and in skin (6, 9, 12, 13).
AC is characterized clinically by life-threatening ventricular arrhythmias and pathologically by acquired and progressive changes with fibro-fatty replacement of the myocardial tissue (14). AC currently comprises several variant patterns: RV, left ventricular (LV) and biventricular involvement (15). AC diagnosis is exceptionally made under the age of 10 years (5, 10, 16). Indeed, in the current case, cardiac abnormalities were not detected at 5 years of age, and the patient remained free of clinical signs of cardiac disease until the age of 10 years. Nevertheless, molecular diagnosis and the consequent in-depth cardiac examinations in a tertiary care centre allowed timely detection of the AC, ICD implantation and appropriate medical treatment initiation. Therefore, the combination of congenital woolly hair and PPK in a young child should always prompt serial cardiac diagnostic work-up, which can be life-saving.
Authors acknowledge support from the Italian Ministry of Health (Ricerca Corrente 2018–2020). IDI-IRCCS and Bambino Gesù Children’s Hospital are healthcare providers of the European Reference Network (ERN)-skin, and Bambino Gesù Children Hospital participates to ERN GUARD HEART.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize