


1University Medical Center Utrecht, Utrecht, The Netherlands, 2Regeneron Pharmaceuticals, Inc., Tarrytown, NY, USA, 3Analysis Group, Inc., New York, NY, USA, 4Sanofi, Chilly-Mazarin, France, and 5Sanofi, Bridgewater, NJ, USA
#Co-last authors.
Dupilumab is approved for uncontrolled moderate-to-severe atopic dermatitis (AD); cyclosporine is approved for severe AD for ≤ 1 year. The efficacy/effectiveness of these treat-ments was compared indirectly. Regression models used pooled patient-level data to estimate response (Eczema Area and Severity Index [EASI] EASI-50/EASI-75 at weeks 12–16 and 24–30) to dupilumab 300 mg every 2 weeks (CHRONOS [NCT02260986]) or cyclosporine (University Medical Center). Models were adjusted for sex, baseline EASI, and thymus and activation-regulated chemokine level. A total of 106 patients received dupilumab (+ topical cortico-steroids; + TCS), and 57 received cyclosporine (+ TCS). Among University Medical Center patients, estimated EASI-50 responders were, dupilumab vs. cyclosporine, 91% vs. 77% (p = 0.038; weeks 12–16), and 96% vs. 67% (p < 0.0001; weeks 24–30); EASI-75 responders were 78% vs. 56% (p = 0.016; weeks 12–16) and 80% vs. 47% (p <0.001; weeks 24–30). Among CHRONOS patients, estimated EASI-50 responders were 90% vs. 74% (p <0.038; weeks 12–16) and 92% vs. 53% (p < 0.0001; weeks 24–30); EASI-75 responders were 75% vs. 52% (p = 0.016; weeks 12–16) and 74% vs. 40% (p <0.001; weeks 24–30), respectively. These results suggest a higher relative efficacy of dupilumab vs. cyclosporine.
Key words: adult; atopic dermatitis; cyclosporine; dupilumab; eczema; Eczema Area and Severity Index.
Accepted May 16, 2019; E-published May 17, 2019
Acta Derm Venereol 2019; XX: XX–XX.
Corr: Marjolein de Bruin-Weller, University Medical Center Utrecht, Heidelberglaan 100, NL-3584 CX Utrecht, The Netherlands. E-mail: m.s.debruin-weller@umcutrecht.nl
Dupilumab is approved for uncontrolled moderate-to-severe atopic dermatitis; cyclosporine is approved for severe atopic dermatitis. Efficacy/effectiveness were compared indirectly using regression models. Estimated response was evaluated using 50% improvement in Eczema Area and Severity Index (EASI-50) and 75% improvement (EASI-75) at weeks 12–16 and 24–30 to dupilumab (data from CHRONOS study) or cyclosporine (data from University Medical Center; UMC). For UMC patients, EASI-50 responders were, dupilumab vs. cyclosporine, 91% vs. 77% at weeks 12–16, and 96% vs. 67% at weeks 24–30; EASI-75 responders were 78% vs. 56% at weeks 12–16, and 80% vs. 47% at weeks 24–30. For CHRONOS, EASI-50 responders were 90% vs. 74% at weeks 12–16 and 92% vs. 53% at weeks 24–30; EASI-75 responders were 75% vs. 52% at weeks 12–16, and 74% vs. 40% at weeks 24–30, respectively. These results suggest a higher relative efficacy of dupilumab vs. cyclosporine.
Atopic dermatitis (AD) is a common chronic inflammatory skin disease, which is prone to disease exacerbations. In adults, the estimated prevalence of AD is between 2% and 5%, depending on region (1). In the majority of patients, AD can be treated adequately with topical agents and/or ultraviolet (UV) light (2). However, in a subpopulation of patients with moderate-to-severe AD, the disease remains inadequately controlled despite these treatments, with patients still experiencing signs (e.g. lesions, redness) and symptoms (e.g. itch, sleep disturbance). For these patients, systemic immunomodulating treatment is indicated. The decision to start systemic medication should be based on the severity of skin lesions; symptoms such as itch, pain, and sleep disturbance; and the impact on quality of life (2).
Cyclosporine A is the only oral immunosuppressant approved for the treatment of severe AD in some European countries and in Japan (3, 4). The clinical efficacy of cyclosporine in moderate-to-severe AD was supported by a systematic review of 14 randomized controlled trials, although no conclusion could be drawn about long-term safety (5). Furthermore, many of the studies were conducted in the early 1990s, and well-validated efficacy outcome measures, such as the Eczema Area and Severity Index (EASI), had not yet been developed. In a more recent study of 356 patients with AD who were receiving long-term treatment with cyclosporine, nearly half of the patients cited lack of efficacy and/or side-effects as their reason for discontinuation of treatment (6). Treatment response to cyclosporine has been met with various levels of success (6, 7). Other immunosuppressant agents, including azathioprine and mycophenolate mofetil, are only recommended for use in adults by the European guidelines if cyclosporine A is either not effective or is contraindicated (8). There remains a high unmet need for efficacious and safe therapeutics for inadequately controlled, moderate-to-severe AD.
Dupilumab is a fully human VelocImmune®-derived (9, 10) monoclonal antibody directed against interleukin (IL)-4 receptor alpha that inhibits signaling of IL-4 and IL-13 cytokines, key drivers of type 2 immune diseases, such as AD, asthma, allergic rhinitis, and eosinophilic esophagitis. Dupilumab is approved in the European Union (EU), USA, Japan, and other countries for treatment of inadequately controlled moderate-to-severe AD in adults. The clinical efficacy and safety of dupilumab ± topical corticosteroids (TCS) have been demonstrated in clinical trials of 16 weeks’ (SOLO 1 & 2) and 52 weeks’ (CHRONOS) duration (11, 12), as well as in patients for whom cyclosporine failed or was contraindicated (CAFÉ; 13).
Both cyclosporine and dupilumab are approved in most European countries for patients whose disease cannot be controlled by, or who are intolerant of, topical treatment (3, 14). However, there is a lack of head-to-head data comparing these 2 agents.
The aim of this study was to assess the relative effectiveness of dupilumab vs. cyclosporine in adult patients with moderate-to-severe AD. This comparison was achieved by estimating the proportions of patients with treatment responses based on improvements from baseline in EASI score of 75% (EASI-75; primary endpoint of CHRONOS) or 50% (EASI-50; secondary endpoint of CHRONOS).
Patient-level data on dupilumab and cyclosporine treatment of AD were obtained from 2 different data sources. Dupilumab data were obtained from the phase 3 trial LIBERTY AD CHRONOS (CHRONOS), the design and results of which have been reported elsewhere (12). CHRONOS was a global, randomized, double-blind, placebo-controlled trial conducted in 14 countries in Europe, Asia-Pacific, and North America between 3 October 2014, and 31 July 2015. Adult patients (aged ≥ 18 years) with moderate-to-severe AD and an inadequate response to medium- or higher-potency TCS treatment were included. The trial evaluated 2 dupilumab dose regimens: 300 mg every week (qw) plus concomitant TCS, 300 mg every 2 weeks (q2w) plus concomitant TCS, or placebo plus TCS. This analysis focused on the dupilumab 300 mg q2w plus TCS treatment arm, the dose regimen approved by the European Medicines Agency. Key inclusion criteria for the CHRONOS study included the presence of AD for ≥ 3 years before screening; a documented history within 6 months before screening of inadequate response to medium-to-high-potency TCS (with or without topical calcineurin inhibitor, as appropriate) or documented systemic treatment within the previous 6 months, or both; and an Investigator’s Global Assessment (IGA) score of ≥ 3 (moderate-to-severe, on a scale of 0–4) and an EASI score of ≥ 16 at screening and baseline (12).
Patient-level data on cyclosporine were obtained from patients treated with cyclosporine in daily practice at the Department of Dermatology and Allergology, University Medical Center (UMC) Utrecht, the Netherlands. SAS enterprise (https://sas.com) was used to extract all patients treated with cyclosporine A between January 2015 and September 2017 in the UMC Utrecht with a diagnosis of “atopic dermatitis”. Most patients were treated with cyclosporine as the first choice of systemic treatment, according to the local treatment protocol. This involved treatment initiation at a high dose, 5 mg/kg/day, for a 3–6-week induction phase, followed by gradual tapering of the dose based on clinical response to a dose of 2–3 mg/kg/day in the maintenance phase. Tapering of the cyclosporine dose was undertaken in all patients to balance the long-term safety/effectiveness profile and to establish the lowest dose at which cyclosporine remained therapeutically effective. This approach reflects how cyclosporine is used in real-world practice, because of its known toxicity profile. Concomitant use of TCS was permitted as needed for all patients treated with cyclosporine.
Baseline data recorded for the UMC Utrecht patients included age, sex, EASI score, and thymus and activation-regulated chemokine (TARC) level at the date of cyclosporine initiation. EASI scores were available at weeks 3, 12–16, and 24–30 after the index date. Data on treatment duration, reason for discontinuation, and the cyclosporine dose were also collected at these time points. Patients treated with cyclosporine were included in the analysis if they had been treated with cyclosporine between January 2015 and September 2017 for a duration of ≥ 3 weeks, and if baseline characteristics (EASI, serum TARC level, sex, and age) and at least one follow-up EASI value were available. Outcomes for the analysis included EASI-50 and EASI-75. Patients reaching the given EASI improvement outcomes (EASI-50 or EASI-75) were defined as “responders,” while those not meeting EASI-50 or EASI-75 were defined as “non-responders.”
Because the cyclosporine population was treated in daily practice without fixed clinic visits, the analysis of EASI-50 and EASI-75 spanned 2 different time periods, between weeks 12 and 16 and between weeks 24 and 30. In contrast, CHRONOS patients were treated in a clinical trial setting, with EASI outcomes assessed at specific time points rather than ranges. To facilitate a comparison with outcomes in the cyclosporine population, EASI-50 and EASI-75 scores for patients in the CHRONOS study are reported here as those obtained at weeks 16 and 28.
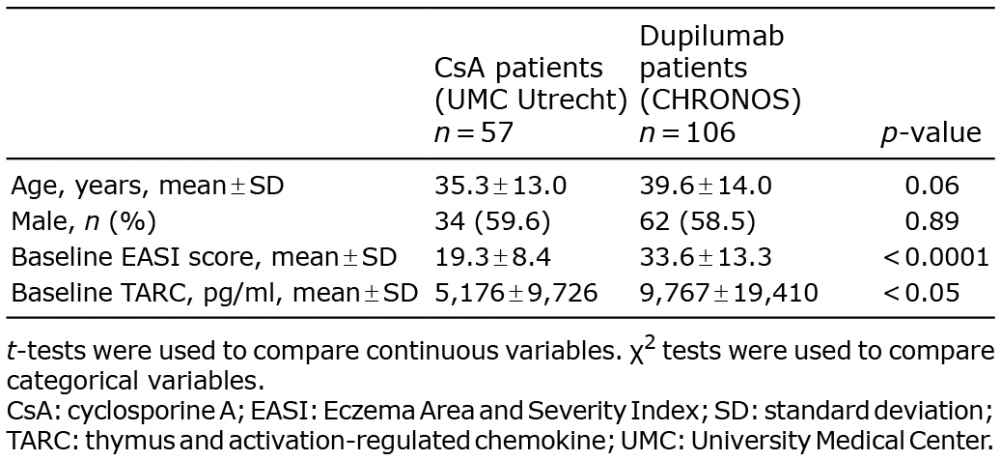
Age, sex, EASI score, and TARC level were available at baseline for both populations. For continuous variables (age, EASI, and TARC), data were presented as means, medians, and standard deviations (SD). For categorical variables, data were presented as frequencies and percentages of patients. The 2 populations were compared using t-tests for continuous variables and χ2 tests for categorical variables. A threshold of p < 0.05 was used to define statistical significance.
Logistic regression analysis was performed to assess the efficacy outcomes for each endpoint. The dependent variable was EASI-50/EASI-75 (achieved or not achieved), and the focal regressor was a treatment indicator for cyclosporine vs. dupilumab use. Missing EASI values were imputed by means of the last observation carried forward (LOCF) method for both treatment populations. The other regressors in the model were sex, baseline EASI, and baseline TARC level, and adjusted-weighting was done according to these baseline data. Patients with missing baseline TARC levels or EASI scores were excluded from the analysis.
Coefficients from the adjusted regression models were used to estimate the mean predicted rate of responders under each treatment scenario (treatment with dupilumab vs. with cyclosporine) for the CHRONOS and UMC Utrecht populations separately. This enabled the prediction of responder rates for dupilumab and cyclosporine within each of the study populations. Standard errors for the estimated proportion of EASI responders were calculated using a bootstrapping technique with re-sampling (number of iterations = 1,000). The variance estimates (i.e. standard errors) were thereby generated instead of under parametric distribution assumptions around the predicted EASI responder rates. p-values for the treatment indicator (dupilumab vs. cyclosporine) in each model were reported.
The relative improvement in efficacy/effectiveness of dupilumab vs. cyclosporine over time between weeks 12–16 and weeks 24–30 was tested statistically with confidence intervals (CI) calculated by a bootstrap method with 1,000 iterations.
A total of 163 patients were included in the analysis. Out of 105 patients in the database at the UMC Utrecht, 48 were excluded from further analysis based on the following exclusion criteria: other treatment indication than atopic dermatitis, treatment duration < 3 weeks, missing baseline characteristic (EASI, serum TARC level, sex, age) and < 1 available follow-up EASI value, leading to 57 treated with cyclosporine + TCS. A total of 106 patients with dupilumab q2w + TCS in CHRONOS were included in the analysis. Of the 57 cyclosporine-treated patients, 40 (70%) had no history of previous treatment with oral immunosuppressive drugs. Of 17 (30%) patients who had previously received one or more immunosuppressive drugs, 6 (35%) had received metho-trexate, 12 (71%) cyclosporine, 3 (18%) azathioprine, and 1 (6%) mycophenolic acid. In contrast, 43 (41%) of the dupilumab-treated patients had previously received non-steroidal immunosuppressants to treat AD. Of these patients, 8 (19%) had received methotrexate, 33 (77%) cyclosporine, 8 (19%) azathioprine, and 8 (19%) mycophenolic acid.
Age and sex did not significantly differ between cyclosporine-treated and dupilumab-treated patients (Table I). Baseline EASI score and baseline serum TARC level were significantly higher in patients treated with dupilumab (EASI: 33.6 ± 13.3, p < 0.0001; TARC: 9,767 ± 19,410, p < 0.05) than in cyclosporine-treated patients (EASI: 19.3 ± 8.4, TARC: 5,176 ± 9,726).
Table I. Baseline characteristics
During the follow-up period of 24–30 weeks, 5 (8.8%) patients discontinued cyclosporine treatment. Reasons for discontinuation were side-effects (3 patients; 5.3%), ineffectiveness (1 patient; 1.8%), and pregnancy (1 patient; 1.8%). The median (interquartile range) cyclosporine dose at the different time points was 4.8 (0.8) mg/kg at baseline, 3.3 (0.7) mg/kg after 12–16 weeks’ treatment, and 3.0 (0.9) mg/kg after 24–30 weeks’ treatment. Treatment characteristics of the patients treated with cyclosporine are shown in Table II.
Of the 106 patients treated with dupilumab, 8 (7.5%) discontinued dupilumab treatment within the follow-up period of 28 weeks. Reasons for discontinuation of treatment were withdrawal by subject (4 patients; 3.8%), physician decision (2 patients; 1.9%), adverse event (1 patient; 0.9%), and protocol violation (1 patient; 0.9%) (Table II).
Table II. Treatment characteristics of the University Medical Center (UMC) Utrecht patients who received cyclosporine and the CHRONOS patients who received dupilumab
Table III shows the adjusted regression-estimated proportions of responders to each treatment for the UMC Utrecht and CHRONOS patients; these data are presented graphically in Figs 1 and 2. Both the UMC Utrecht and CHRONOS patients had a higher estimated proportion of EASI responders with dupilumab than with cyclosporine treatment. Among UMC Utrecht patients, the estimated proportions of EASI-50 responders to dupilumab vs. cyclosporine treatment were, respectively, 91% vs. 77% (p = 0.038) in weeks 12–16 and 96% vs. 67% (p < 0.0001) in weeks 24–30; the estimated proportions of EASI-75 responders were 78% vs. 56% (p = 0.016) in weeks 12–16 and 80% vs. 47% (p < 0.001) in weeks 24–30. Among the CHRONOS trial patients, the estimated proportions of EASI-50 responders to dupilumab vs. cyclosporine treatment were, respectively, 90% vs. 74% (p < 0.038) in weeks 12–16 and 92% vs. 53% (p < 0.0001) in weeks 24–30; the estimated proportions of EASI-75 responders were 75% vs. 52% (p = 0.016) in weeks 12–16 and 74% vs. 40% (p < 0.001) in weeks 24–30. For all outcome measures at all time points, the actual (observed) percentages of patients who responded to cyclosporine in the UMC Utrecht study were the same as those estimated from the model. Likewise, in the CHRONOS study, the observed proportions of patients responding to dupilumab were identical to the estimated proportion of responders (Table III).
Table III. Adjusted regression results: estimating proportions of treatment responders to dupilumab and cyclosporine based on EASI improvement
Fig. 1. Estimated proportions of Eczema Area and Severity Index EASI. (A) EASI-50 and (B) EASI-75 responders in the University Medical Center (UMC) Utrecht population (n = 57). Solid colour represents observed values. Hatched colours represent the estimated values. SE: standard error.
Fig. 2. Estimated proportions of Eczema Area and Severity Index (EASI). (A) EASI-50 and (B) EASI-75 responders in the CHRONOS population (n = 106). Solid colour represents observed values. Hatched colours represent the estimated values. SE: standard error.
According to the EASI-50 criterion, the differences in responder rates of dupilumab vs. cyclosporine treatment during weeks 12–16 were 14% (UMC Utrecht population) and 16% (CHRONOS population). By weeks 24–30, these differences had increased to 29% (UMC Utrecht population) and 39% (CHRONOS population). A similar trend in treatment difference was observed for EASI-75. The EASI-75 responder rates for dupilumab vs. cyclosporine treatment during weeks 12–16 differed by 22% (UMC Utrecht population) and 23% (CHRONOS population). By weeks 24–30, these differences had increased to 33% (UMC Utrecht population) and 34% (CHRONOS population).
The proportion of EASI-50 responders to dupilumab increased slightly between weeks 12–16 and weeks 24–30 in the UMC Utrecht population (from 91% to 96%) and the CHRONOS population (from 90% to 92%). In contrast, the proportion of EASI-50 responders to cyclosporine decreased between weeks 12–16 and weeks 24–30 in the UMC Utrecht population (from 77% to 67%) and the CHRONOS population (from 74% to 53%). The proportion of EASI-75 responders to dupilumab increased slightly or remained stable between weeks 12–16 and weeks 24–30 in both the UMC Utrecht (from 78% to 80%) and CHRONOS (from 75% to 74%) populations. The proportion of patients with EASI-75 responders to cyclosporine decreased between weeks 12–16 and weeks 24–30 in both the UMC Utrecht (from 56% to 47%) and CHRONOS (from 52% to 40%) populations.
The relative increase in the proportion of EASI-50 responders to dupilumab vs. cyclosporine from weeks 12–16 to weeks 24–30 was statistically significant in both the UMC Utrecht (15%; 95% CI 2%, 29%) and the CHRONOS (23%; 95% CI 5%, 40%) populations (Table IV). However, the change in proportion of EASI-75 responders to dupilumab vs. cyclosporine from weeks 12–16 to weeks 24–30 was not statistically significant in either the UMC Utrecht (11%; 95% CI −3%, 25%) or the CHRONOS (12%; 95% CI −4%, 26%) populations.
Table IV. Adjusted regression results: relative differences in proportions of treatment responders to dupilumab vs. cyclosporine over time
This analysis suggests a higher relative efficacy of dupilumab compared with cyclosporine effectiveness in the treatment of moderate-to-severe AD, as assessed by 50% and 75% improvements in patients’ EASI scores. A direct-comparison (head-to-head) trial between dupilumab and cyclosporine for the treatment of moderate-to-severe AD would be the most rigorous means of comparing the clinical efficacy and safety of both therapies. No such published data were available. A systematic review of the literature suggested that, in the few trials assessing the efficacy of cyclosporine in AD, sample size is generally lower than 30, EASI is often not considered, and outcomes available are often reported at much earlier time points (4–8 weeks) than in dupilumab trials. The lack of adequate evidence and “networks” for comparison of dupilumab vs. cyclosporine efficacy based on published trials precluded us from using the conventional indirect comparison approach of network meta-analysis. Hence, we used patient-level data to conduct an indirect comparison of the efficacy of the 2 agents in the treatment of AD.
The relative efficacy of dupilumab vs. efficacy/effectiveness of cyclosporine improved significantly over time, on the basis of the EASI-50 response. Patients who received cyclosporine were treated, according to a defined protocol, with a high starting dose (5 mg/kg/day) over 3–6 weeks and a stepwise tapering to a maintenance dose of 2–3 mg/kg/day. The cyclosporine dose was adjusted on the basis of individual factors, including effectiveness and side-effects. Tapering of the dose reflects real-world practice and is required to avoid unwanted side-effects, while maintaining a dose with sufficient clinical effectiveness over a prolonged treatment period. Most patients have an adequate response to a high dose, but may relapse after dose reduction. Because the dose of cyclosporine is tapered stepwise, the effect of the high starting dose may continue until the first time point, weeks 12–16. Patients treated with dupilumab, on the other hand, were given 300 mg dupilumab q2w without dose adjustment. The dose reduction after the 3–6 week induction phase in patients treated with cyclosporine may have contributed to the trend in improvement in relative efficacy/effectiveness that was observed for dupilumab vs. cyclosporine over time. However, it should be noted that clinicians at UMC Utrecht are highly experienced with cyclosporine treatment and the need to taper the dose on the basis of balancing maximal effectiveness while limiting safety issues.
Blauvelt et al. (12) reported a lower week 16 EASI-50 response rate (80%) for the dupilumab q2w arm (post hoc analysis) compared with the 90% response rate reported in this study (Table III). This difference can be explained by the use of a highly conservative non-responder imputation in the CHRONOS analysis reported by Blauvelt et al. (12). In that publication, patients were defined as non-responders in cases of missing EASI values and after rescue treatment initiation or study withdrawal. In the present analysis, a LOCF analysis was performed in cases of missing follow-up EASI values. The analyses used patient-level data, which enabled select baseline characteristics to be adjusted in the regression models.
Adjustment in the multivariate regression models was possible only for available baseline characteristics that were common to both study populations, and the models did not adjust for unknown or unmeasured characteristics. Because patients were from different settings it is possible that unobserved confounders may exist that are not accounted for in the model.
The efficacy of cyclosporine in published clinical trials of AD has been reported at much earlier time points than the primary endpoint (16 weeks) in dupilumab trials (15, 16). Also, none of the cyclosporine trials used EASI as an endpoint, which limited the ability to perform a network meta-analysis or matching-adjusted indirect comparison of the efficacy of cyclosporine with an emergent systemic therapy, such as dupilumab. However, it should be noted that the proportions of patients with EASI-50 (51%) and EASI-75 (34%) after 3–6 months’ treatment with cyclosporine A in 35 patients from the German Atopic Eczema Registry (17) are lower than that observed in our study, although mean baseline EASI values were similar.
Safety was not the primary objective of this comparison, and is a difficult factor to compare in a retrospective design. Furthermore, the proactive recording of safety signals in a clinical trial setting is more rigorous than the spontaneous nature of safety reporting typical in daily practice. Therefore, adverse events were not reported in this analysis. To provide some measure of comparison, the reasons for treatment discontinuation due to side-effects have been reported for each population.
There are a number of limitations to the current study, including the fact that, since patients were not randomized, causality cannot be inferred from the findings. In addition, the overall sample size was relatively small for the purposes of an indirect comparison, and replication of the analysis with a larger sample may be warranted. Furthermore, as the data represent a convenience sample from already-collected data, no power calculations were conducted a priori, which may have increased the likelihood of statistical type 2 errors. Regarding the logistic regression model, it is also possible that all relevant predictors of treatment response may not have been included.
The differences between the 2 population types can also be considered a limitation: CHRONOS was a global randomized controlled trial, whereas the UMC Utrecht study was conducted at one local site. It is therefore possible that unobserved differences in practice patterns or patient characteristics may have contributed to some of the study findings. Furthermore, patients participating in clinical trials are usually screened on the basis of precise inclusion and exclusion criteria, and therefore have similar characteristics. However, patients treated in daily practice often differ in characteristics such as comorbidities and medication use. In a real-life setting the balance between effectiveness and side-effects determines whether treatment will be continued or dose adjustment is necessary. Differences in patient characteristics and dosage adjustment based on clinical characteristics and effectiveness in the patients treated with cyclosporine might therefore have influenced the results. Disease history was not verified independently of the patients’ self-report and is not reported here due to a high chance of recall bias. A final limitation of the study concerns collection of the data. The time point at which EASI was reported in patients treated with cyclosporine spanned a multi-week interval, as would be anticipated in daily practice, so there was no granularity in the exact timing of its assessment. By contrast, data from the CHRONOS patients were taken from a specific assessment point, as specified in the clinical trial protocol.
In conclusion, despite the several inherent limitations of an indirect comparison, our findings suggest that dupilumab has greater relative efficacy than cyclosporine in the treatment of moderate-to-severe AD in adult patients, as captured using a well-validated outcome measure, improvement in EASI score. Furthermore, the relative efficacy benefit in favor of dupilumab for EASI-50 increased over time.
This research was sponsored by Sanofi and Regeneron Pharmaceuticals, Inc. ClinicalTrials.gov Identifier: NCT02260986. Medical writing/editorial assistance was provided by Juliet H. A. Bell, PhD, of Excerpta Medica, funded by Sanofi Genzyme and Regeneron Pharmaceuticals, Inc. Review and input on statistical methods for indirect comparison was provided by Yingxin Xu, Regeneron Pharmaceuticals Inc.
Institutional Review Board (IRB) Approval Statement. The CHRONOS study was conducted in accordance with the provisions of the Declaration of Helsinki, International Conference on Harmonisation Good Clinical Practice guidelines (version R1), and applicable regulatory requirements. All patients provided signed written informed consent. The protocol and all relevant study forms of the CHRONOS study were approved by all relevant institutional review boards and an independent ethics committee. An independent data monitoring committee monitored patient safety.
Regarding the UMC Utrecht study, the Medical Research Ethics Committee confirmed that the Medical Research Involving Human Subjects Act (WMO) does not apply to the above-mentioned study and therefore an official approval of this study by the Medical Ethics Research Committee of UMC Utrecht was not required under the 1999 Medical Research Involving Human Subjects Act.
Conflicts of interest: LFMA: nothing to disclose. AG, AK, ZC, YL, NMHG: Regeneron Pharmaceuticals, Inc. − employees and shareholders. HvO-M: Sanofi – advisory board member. RA, ET: Analysis Group, Inc. – employees; Analysis Group, Inc. has received consultancy fees from Regeneron Pharmaceuticals, Inc. and Sanofi. GB-LB, ER, GP, LE: Sanofi − employees, may hold stock and/or stock options in the company. MdB-W: Regeneron Pharmaceuticals, Inc., Sanofi Genzyme – principal investigator, advisory board member, consultant; AbbVie – principal investigator, advisory board member; Pfizer – principal investigator, advisory board member; Eli Lilly, UCB – advisory board member.


 Click to show fullsize
Click to show fullsize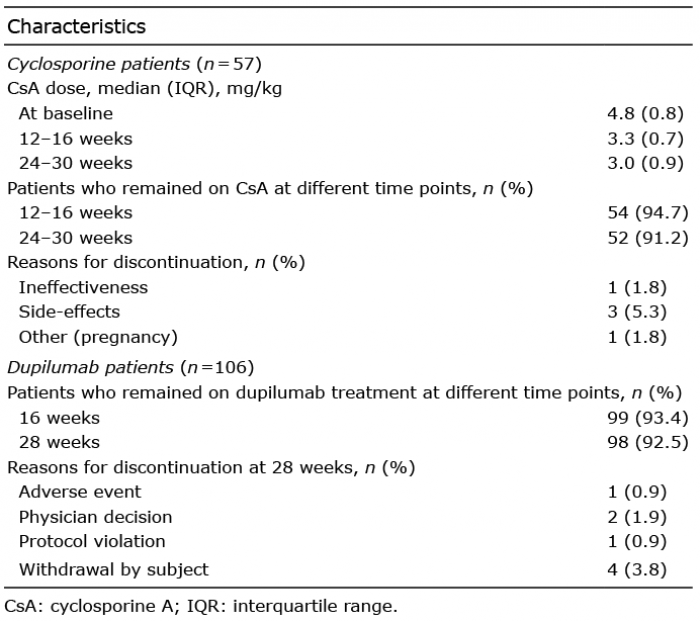 Click to show fullsize
Click to show fullsize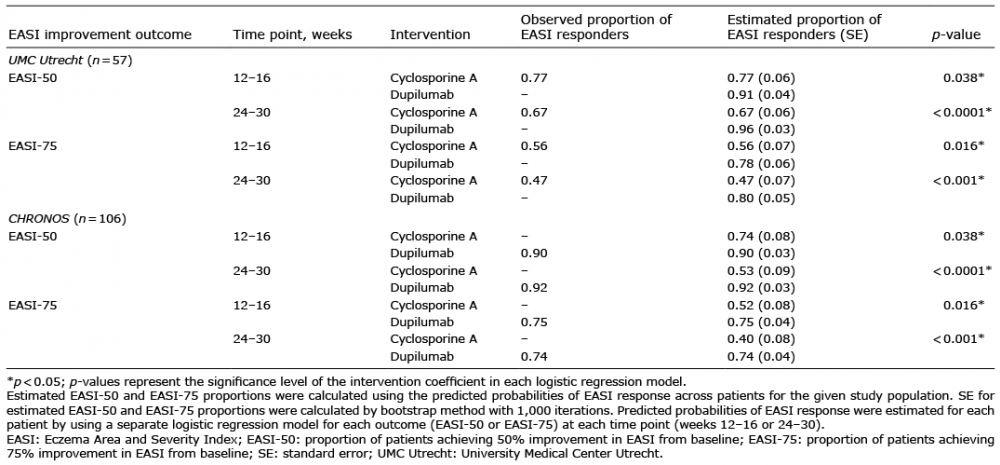 Click to show fullsize
Click to show fullsize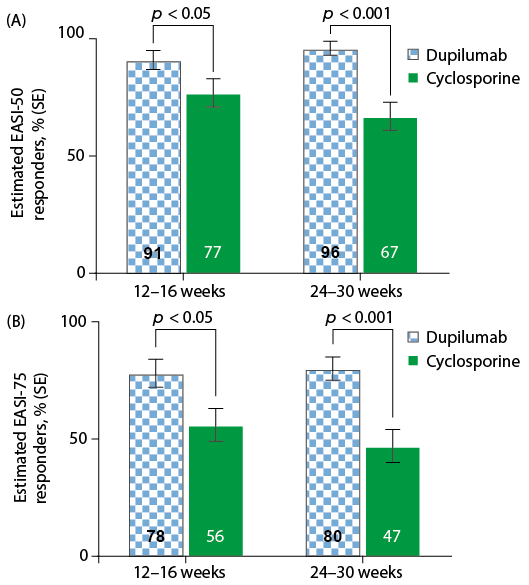 Click to show fullsize
Click to show fullsize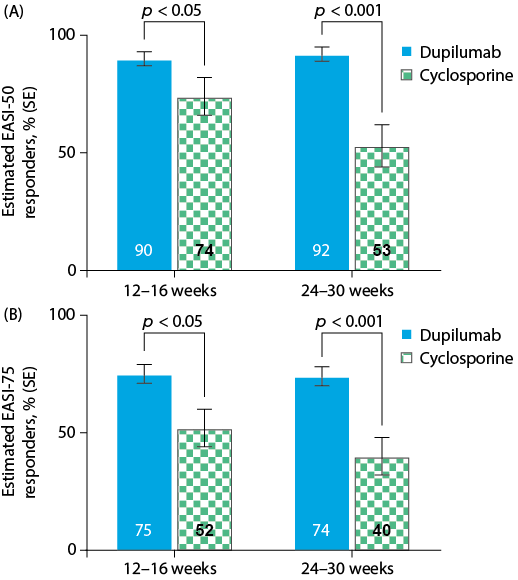 Click to show fullsize
Click to show fullsize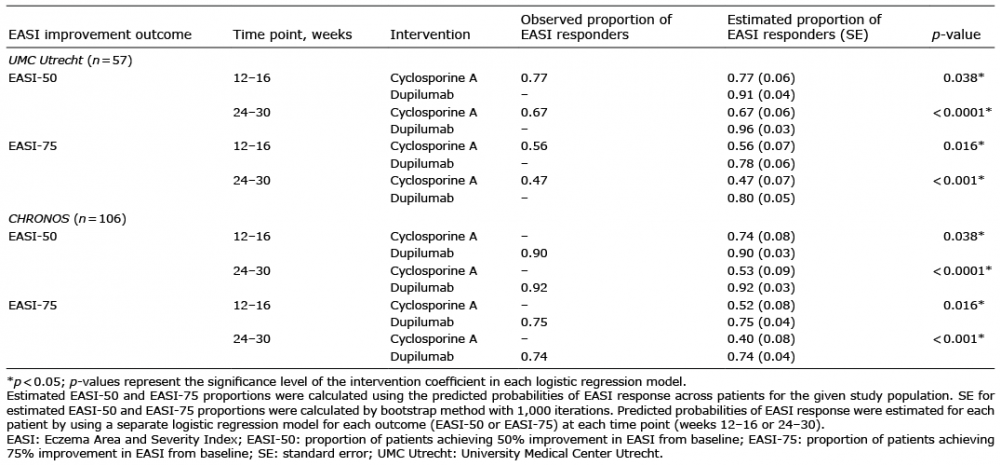 Click to show fullsize
Click to show fullsize