


1Department of Dermatology, 2Department of Pathology, 3Catholic University Lymphoma Group, 4Department of Hematology, Seoul St Mary’s Hospital, College of Medicine, The Catholic University of Korea, 5Department of Dermatology, Uijeongbu St Mary’s Hospital, College of Medicine, The Catholic University of Korea, 6Department of Dermatology, St Vincent’s Hospital, College of Medicine, The Catholic University of Korea, and 7Department of Dermatology, Yeouido St Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, Korea
Castleman’s disease is a rare disease of the lymph nodes and related tissues, presenting as angiofollicular or giant lymph node hyperplasia. Although various skin manifestations have been reported to occur in Castleman’s disease, a comprehensive study of cutaneous disorders in Castleman’s disease is lacking. Therefore, the aim of this study was to investigate Castleman’s disease-associated cutaneous disorders. The medical records of 57 patients with Castleman’s disease who visited our hospitals from January 2007 to May 2018 were analysed retrospectively. Patients were classified according to the presence of skin involvement. Plasma variant-type Castleman’s disease and multicentric Castleman’s disease were more commonly found in patients with Castleman’s disease with a cutaneous disorder than in those without a cutaneous disorder. In addition, the skin disorders were classified according to pathomechanisms: immune complex-related (paraneoplastic pemphigus, xanthogranulomas), cytokine-related (vasculitis-like lesion, cherry angioma, hyperpigmentation), and non-specific (pruritus). This study builds on previous case reports of cutaneous disorders in Castleman’s disease and proposes a new classification system.
Key words: Castleman’s disease; interleukin-6; lymphoproliferative disorder; paraneoplastic itch; POEMS syndrome.
Accepted Jun 28, 2019; E-published Jul 8, 2019
Acta Derm Venereol 2019; XX: XX–XX.
Corr: Ji Hyun Lee, Department of Dermatology, Seoul St Mary’s Hospital, College of Medicine, The Catholic University of Korea, 222 Banpo-daero, Seocho-gu, Seoul 06591, Korea. E-mail: ejee@catholic.ac.kr
Castleman’s disease is a rare lymphoproliferative disorder, which is often accompanied by various cutaneous manifestations. However, Castleman’s disease-related cutaneous manifestations have not yet been systematically reviewed. The aim of this study was to examine the clinical, histopathological and laboratory characteristics of cutaneous manifestations in Castleman’s disease, through a retrospective review of medical records. The study aimed to provide a comprehensive understanding of Castleman’s disease-associated cutaneous disorders by subclassifying cutaneous disorders according to their underlying pathomechanisms. The study highlights the role of dermatologists in an in-depth review of various skin disorders related to Castleman’s disease.
Benjamin Castleman and co-workers were the first to describe Castleman’s disease (CD), in 1956 (1). CD is a rare polyclonal lymphoproliferative disorder, also known as angiofollicular lymphoid hyperplasia or giant lymph node hyperplasia (1). There are 2 clinical types of CD: (i) unicentric CD (UCD) and (ii) multicentric CD (MCD). UCD manifests as a localized enlargement of mainly mediastinal or abdominal lymph nodes, often with pressure symptoms. MCD most commonly manifests as generalized lymph node enlargement, affecting multiple compartments throughout the neck, chest, abdomen and pelvis, with various systemic symptoms, and has a poorer clinical prognosis than that for UCD (2). MCD can be further divided based on Kaposi sarcoma (KS)-associated herpesvirus/human herpesvirus-8 (HHV-8) infection status. Furthermore, HHV-negative MCD can be classified into polyneuropathy-organomegaly-endocrinopathy-myeloma protein-skin changes syndrome (POEMS)-associated MCD and idiopathic MCD.
The pathogenesis of CD is not yet fully understood. Fajgenbaum & Shilling (3) attempted to classify the pathogenesis according to the CD subtype. Viral, neoplastic, and reactive inflammatory mechanisms have all been proposed as aetiological mechanisms in UCD. In some cases of UCD (4), tests for the Epstein-Barr virus yielded positive results. In HHV-8-associated MCD, the HIV infection or immune-deficient condition is thought to drive the clinicopathological symptoms of CD. In addition, an increased level of vascular endothelial growth factor (VEGF) or activation of other factors by viral G-protein coupled receptors may be involved in HHV-8-associated MCD pathology (5), and increased human interleukin (IL)-6 and viral IL-6 may drive B-cell proliferation or related symptoms. In POEMS-related MCD, VEGF, IL-6, IL-12, transforming growth factor-1β (TGF-1β), and tumour necrosis factor-α (TNF-α) may be induced by the production of mediators from monoclonal plasma cells (6). In idiopathic MCD, elevated levels of IL-6 are also observed, which may be related to disease flares (7).
Skin manifestations, including paraneoplastic pemphigus (PNP), POEMS, xanthoma, KS, etc., are known to occur in up to 55% of patients with CD. However, the previous studies are all case reports and no comprehensive clinical study has been conducted (8). Therefore, in the present study, the clinical, histopathological, and laboratory characteristics of CD were reviewed, with a focus on cutaneous manifestations.
The medical records of all patients who visited Seoul St Mary’s Hospital, Uijeongbu St Mary’s Hospital, St Vincent’s Hospital, and Yeouido St Mary’s Hospital from 1 January 2007 to 31 May 2018 were retrospectively reviewed. A total of 57 patients diagnosed with CD were identified. Of these, 10 patients (17.54%) visited the dermatology department because of a skin disorder. The clinical records of all patients with CD were examined regardless of an accompanying cutaneous disorder.
The following data were collected: age, sex, histopathological type, HIV status, and laboratory blood tests for the erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level. Pathological subtyping was conducted by biopsy.
This study was approved by the ethics committee of the Catholic Medical Center Office of Human Research Protection Program (XC18REDI0021).
Characteristics of study population
Of the 57 patients with CD, 47 did not have a cutaneous disorder and 10 had a cutaneous disorder associated with CD. The patients were first grouped according to the existence of a cutaneous disorder. The group without a cutaneous disorder (mean age 41.8 years; range 3–69 years) had a female predominance, with a male-to-female ratio of 1:1.14. The group with cutaneous disorders (mean age 52.1 years; range 34–76 years) had a male predominance, with a male-to-female ratio of 1.5:1. Clinically, MCD was predominant regardless of the existence of a cutaneous disorder. Among the patients without a cutaneous disorder, 27.7% (n = 13) had UCD and 72.3% (n = 34) had MCD. Among patients with a cutaneous disorder, 20% (n = 2) had UCD and 80% (n = 8) had MCD. Histopathologically, 38.3% of patients without a cutaneous disorder had hyaline vascular variant (HV)-type CD and 38.3% had plasma variant (PV)-type CD. In contrast, 60% of patients with a cutaneous disorder had PV-type and 40% had HV-type CD.
Table I shows the observed CD-associated cutaneous disorders classified according to the possible pathogenesis: immune complex-related manifestations, such as paraneoplastic pemphigus (PNP) (Fig. 1a) and xanthogranulomas (Fig. 1b, c); cytokine-related manifestations, such as vasculitis-like lesions, cherry angiomas, and hyperpigmentation (Fig. 2); and non-specific manifestations, such as paraneoplastic itch. None of the patients were positive for HIV, and the histopathological and immunohistochemical results for HHV-8 in the skin specimens were all negative. In addition, no cases of KS were observed. Nine of the 10 patients underwent laboratory testing; 88.8% (n = 8) had increased ESR or CRP levels (3 patients showed both elevated ESR and CRP, 2 patients showed elevated CRP, and 3 patients showed elevated ESR).
Table I. Sub-classification of cutaneous disorders associated with Castleman’s disease
Fig. 1. Clinical manifestations of immunological-related skin disorders in Castleman’s disease. (a) Paraneoplastic pemphigus presenting as a tender recalcitrant oral ulcer and multiple lichenoid hyperkeratotic plaques on the entire body for 10 months. (b) Solitary xanthogranuloma on the left forearm. (c) Solitary xanthogranuloma on the left thigh.
Fig. 2. Clinical manifestations of cytokine-related skin disorders in Castleman’s disease. (a, b) Multiple cherry angiomas on the trunk. (c) Vasculitis of both lower legs. (d) Hyperpigmentation on the neck.
The observed CD-associated cutaneous manifestations are described in detail below. Three patients had vasculitis and paraneoplastic itch, 2 patients had xanthogranuloma and cherry angioma, and one patient had PNP and hyperpigmentation. In addition, previous reports of cutaneous manifestations associated with CD are summarized in Table II to provide a comprehensive review of the cutaneous disorders associated with CD.
Table II. Previous studies of cutaneous disorders associated with Castleman’s disease
Paraneoplastic pemphigus. One case of PNP presented as a tender, recalcitrant oral ulcer and multiple lichenoid plaques on the entire body. The primary CD lesion and suprarenal mass were excised, and the patient showed excellent response with daily prednisolone (10 mg) and weekly methotrexate (10 mg).
Xanthogranuloma. Two cases of xanthogranuloma presented as single lesions. The xanthogranulomas were resected, and no new lesions have occurred.
Vasculitis-like lesion. Two cases of vasculitis-like lesion presented as painful, multiple, raised red spots on the lower legs, without arthralgia or abdominal pain. One case of vasculitis-like lesion presented as multiple, tiny petechial papules on the entire body during chemotherapy for CD, which showed spontaneous regression after several weeks. All cases occurred after the diagnosis of CD. Patients denied biopsy, but clinical findings strongly suggested vasculitis. The patients showed recurrent progress with topical steroids.
Angioma. Two cases of angioma developed suddenly several years after the diagnosis of CD. No treatment, only follow-up observation, was performed.
Hyperpigmentation. A case of hyperpigmentation was referred to the dermatology department for a differential diagnosis of POEMS syndrome due to hyperpigmentation. The patient was previously diagnosed with CD. A diagnosis of POEMS syndrome could not be made, as the patient did not meet POEMS syndrome mandatory criteria, such as polyneuropathy and monoclonal gammopathy. Rather, she presented with polyclonal gammopathy on an immunofixation test. She also showed no other minor criteria of the skin changes in POEMS syndrome, such as hypertrichosis, apparent skin thickening, angioma, and telangiectasia.
Pruritus. All 3 patients with pruritus had no underlying disease that could cause itching other than CD. The patients were treated with oral antihistamine and topical steroids, and showed a progressive course of improvement and deterioration.
The present study is unique in that it comprised a comprehensive retrospective review of CD-associated cutaneous disorders. Furthermore, the study newly classified the cutaneous disorders according to the pathomechanism (immune complex-related, cytokine-related, or non-specific). In addition, a summary of CD-associated cutaneous disorders in previous reports was provided to further improve our understanding of cutaneous disorders in CD.
In the present study, the mean age of the patients with CD without a cutaneous disorder was 41.8 years, while that of the patients with a cutaneous disorder was 52.1 years. In Talat & Schulte (9) and Zhang et al. (10), the median age of the patients with CD was 37 years. Thus, the patients with CD with a cutaneous disorder in the present study were much older than those without a cutaneous disorder, by approximately 10.3 years, and those in previous reports on CD, by approximately 15 years. Furthermore, in Zhang et al. (10) 65.4% of patients with CD were classified as UCD and 34.6% were classified as MCD. This is inconsistent with the present study, in which MCD was more common than UCD. Because MCD is more strongly associated with systemic symptoms than is UCD, MCD may be more commonly observed in patients who visit a university hospital. Regardless of this potential bias, patients with CD with a cutaneous disorder had a much higher dominance of MCD over UCD than that in patients without a cutaneous disorder. Furthermore, Melikyan et al. (11) reported that the ratio of histological HV-type to PV-type CD was 1:1 among 76 patients with CD, which is the same as that in the patients without a cutaneous disorder in the present study. In contrast, PV-type was dominant in the patients with a cutaneous disorder, comprising 60% of this group. Thus, MCD predominated clinically and PV-type CD predominated pathologically in patients with CD with a cutaneous disorder, unlike that in patients with CD without a cutaneous disorder.
IL-6 is a key mediator in the pathogenesis of MCD. Yabuhara & Komiyama (12) reported that the overproduction of IL-6 by hyperplastic lymph nodes in CD induces both immunoglobulin (Ig) and acute phase reactant production. In the present study, 88.8% of the patients with CD with a cutaneous disorder had increased ESR or CRP levels. PV-type CD and MCD show more frequent systemic complications than do HV-type CD and UCD due to increased IL-6 (8). In the present study, PV-type CD and MCD were more commonly found in patients with CD with a cutaneous disorder than in those without a cutaneous disorder, consistent with previous reports showing that IL-6 plays a key role in the systemic complexity of CD.
Previous studies reported the following as CD-associated cutaneous disorders: PNP (13), lichenoid eruptions (14), nodular eruptions (8), maculopapular eruptions (8), KS (15), autoimmune bleeding disorders, POEMS syndrome (16), cutaneous necrotizing vasculitis, and plane xanthoma (8). PNP is the most well-known CD-associated cutaneous disorder (17–26). UCD is one of the most common causes of PNP, along with non-Hodgkin lymphoma and other haematological neoplasms (27). The patient with PNP in the present study had UCD, consistent with previous reports. Oh et al. (21) and Maier et al. (24) suggested that autoantibodies, such as p200 proteins, Dsg1, Dsg3, and BP180, are involved in the pathogenesis of PNP. Furthermore, Sherman et al. (8) postulated that xanthoma and CD share pathological mechanisms, including enhanced tissue uptake and increased lipolysis caused by the interaction of triglyceride-rich lipoprotein antibodies in the IgG-low density lipoprotein (LDL) complex with macrophage scavenger receptors that results in the formation of xanthomas and deposition of antibody-lipoprotein complexes. Szalat et al. (28) reported that necrobiotic xanthogranuloma is characterized by impaired lipid homeostasis, and is associated with a systemic inflammatory profile including increased CRP and other ILs. However, the patients with xanthoma (n = 2) in the present study all showed normal lipid profiles and normal CRP levels, which ruled out necrobiotic xanthogranuloma. Given the above-noted previous research, we classified PNP and xanthoma as “immunological (antibody)-related” cutaneous manifestations.
In the present study, vasculitis-like lesions, angiomas, and hyperpigmentation present in 3, 2, and 1 patient, respectively. These cutaneous manifestations are minor criteria of the skin changes in POEMS syndrome; however, a diagnosis of POEMS syndrome cannot be made by minor criteria alone. POEMS-associated MCD is thought to be caused by cytokine production from monoclonal plasma cells that have undergone genomic events, such as translocations or deletions. IL-6, IL-12, TGF-1ß, and TNF-α are the main cytokines proposed to drive POEMS symptoms (29). Oshima et al. (30) suggested that the overproduction of IL-6 may contribute to vasculitis as well as the pathogenesis of CD. Furthermore, increased IL-6 in MCD causes B cell proliferation and VEGF secretion, which is critical to the pathogenesis of haemangiomas (i.e. the benign cutaneous proliferation of capillaries) (31). IL-1β is associated with cutaneous pigmentation due to the activation of the pro-opiomelanocortin gene (16). IL-6, VEGF, IL-1β are known to play key roles in both POEMS syndrome itself and the skin changes in POEMS syndrome physiology (16, 31). Therefore, we sub-classified the 5 cases of vasculitis, angioma, hyperpigmentation into the same category based on their “cytokine-related” mechanisms.
Paraneoplastic itch is defined as pruritus that occurs before or during the natural evolution of a haematological disease, most commonly caused by lymphoproliferative malignancies (32). Three patients with CD had pruritus in the present study, without any other systemic disorders related to pruritus. Konda et al. (33) reported that serum serotonin and serum IL-6 levels are significantly correlated with the severity of pruritus in patients with prurigo nodularis. Pruritus directly associated with CD has not yet been reported and its exact pathomechanism has not been identified. However, we propose that pruritus associated with CD could be viewed as a skin manifestation of CD.
An association between KS and CD has been established (5); however, KS was not observed in the present study. Lee et al. (34) reported only 5 cases of KS among 950 HIV-infected patients in Korea from 2000 to 2014, demonstrating the very low incidence of KS in Korea. Furthermore, HHV-8 and HIV infection are known to drive symptoms of CD; however, Han et al. (35) reported that the mean seroprevalence rate of HHV-8 in Korean children was 3.5%. In addition, unlike Mediterranean regions and Africa, the USA, northern Europe, and other Asian countries show a low prevalence of HHV-8. In Korea, there have been very few cases of HIV-related KS, and thus, the development of KS and HIV infections in patients with CD is considered to be low. As the occurrence of KS and HIV/HHV-8 infection in patients with CD seems to be racially and geographically different, further study is needed.
The present study has some limitations. The retrospective method of the study might have introduced some statistical bias. In addition, the sample size of this study was small due to the rarity of CD itself.
The present study is the first to summarize the cutaneous disorders associated with CD. To facilitate a comprehensive understanding, the present study provided a sub-classification for the cutaneous disorders in CD according to their underlying pathomechanism. Furthermore, various immune responses, especially cytokines, such as IL-6, seem to be involved in both CD itself and the cutaneous disorders.


 Click to show fullsize
Click to show fullsize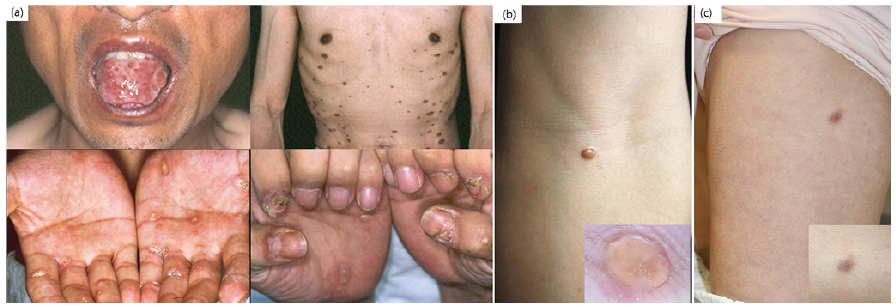 Click to show fullsize
Click to show fullsize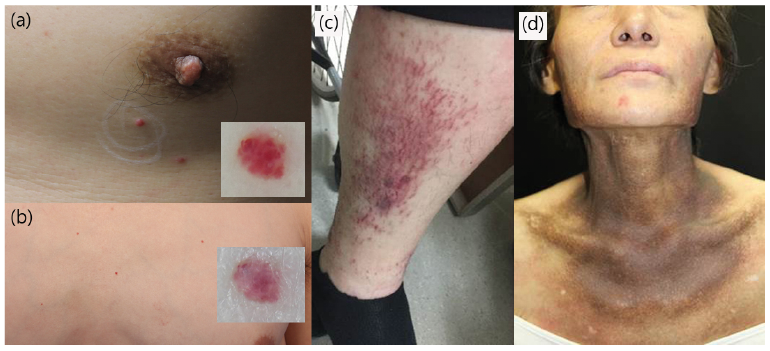 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize