


1Section of Dermatology, Department of Clinical and Experimental Medicine and 2Department of Pathology, University of Parma, Via Gramsci 14, IT-43126 Parma, and 3Dermatology and Venereology, University G. Marconi, Rome, Italy. *E-mail: mariabeatrice.bertolani@gmail.com
*These authors contributed equally to this work.
Accepted Jul 17, 2019; E-published Jul 17, 2019
BRCA1-associated protein-1 (BAP1) tumour predisposition syndrome (TPDS) is a hereditary tumour syndrome caused by germline pathogenic variants in BAP1 (1). It was described for the first time in 2011 by 3 different research groups studying uveal melanoma (UM), mesothelioma (MMe) and cutaneous melanoma (CM). Thereafter, renal cell carcinoma (RCC) was also shown to be associated with the syndrome (2). With regards to the skin, typical mutated lesions “BAP1 inactivated melanocytic tumours” (BIMTs) characterized by a deficiency in BAP1, have been described as part of the syndrome (3). Although the complete phenotype of the syndrome is still being defined, the literature suggests that several other cancers may also be associated with germline BAP1 mutations (2).
A 35-year-old woman was admitted to our Dermatologic Unit (Parma, Italy) for a routine mole evaluation. She was Fitzpatrick’s phototype III with several (> 50) brownish moles and significant photodamage; she had no other dermatological issues. Her family history was significant for cancers: her maternal great-grandmother had breast cancer at the age of 50, her maternal grandmother died from an uveal melanoma at the age of 47, and her mother had breast cancer and basal cell carcinoma (BCC), both occurring in her 50s.
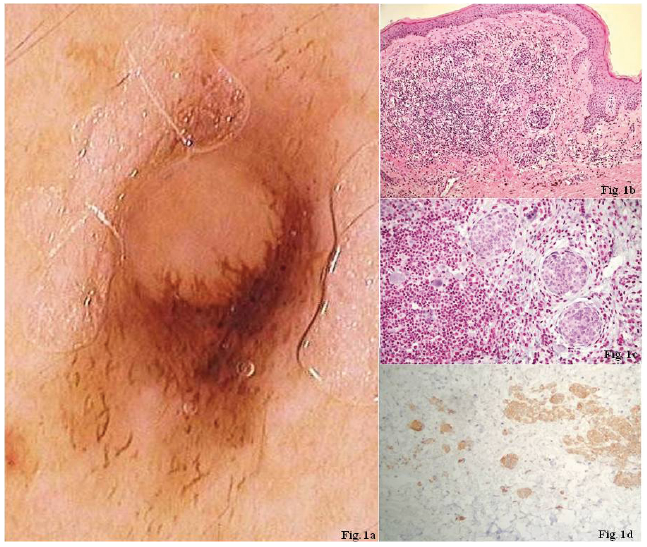
Clinical examination revealed the presence of an asymptomatic pink and light brown, dome-shaped papule on the left hip. Dermoscopic evaluation showed an atypical ridge extending through a pink central dermal area with the presence of atypical peripheral polymorphic vessels (Fig. 1a); therefore the lesion was excised. Histologically, it was characterized by a compound naevus associated with an intradermal component of variably-sized spitzoid melanocytes surrounded by lymphocytes; mitoses were absent (Fig. 1b). Suspicion of BIMT should occur if the presence of a ”spitzoid” appearance and lymphoid infiltrate is identified in an otherwise usual melanocytic naevus. In the nodule, dermal nests of epithelioid melanocytes with abundant eosinophilic cytoplasm, oval nuclei and moderate pleomorphism, are permeated by lympho-cytic infiltrates. In this patient, the spitzoid cells were negative for BAP1 and positive for p-16 and BRAF-v600E mutations (Fig. 1c, d), leading to a diagnosis of an atypical Spitz naevus with loss of the BAP1 protein, associated with a melanocytic compound naevus. As recommended by the literature a wide local excision was performed (4).
The patient underwent further investigations due to the suspicion of a BAP1-TPDS: fundoscopy revealed the presence of a naevus of the iris with no pathological features; a total-body CT scan showed a 2-cm solid exophytic mass in the right kidney which enhanced with contrast; a PET scan was negative; MRI scan confirmed the presence of a solid mass in the right kidney (14 × 15 mm) that was excised with diagnosis of angiomyolipoma; immunohistochemistry revealed cells positive for BAP1, leading to the conclusion that this lesion was unlikely to be related to the syndrome. A similar smaller contralateral lesion (10 × 11 mm) remains under surveillance. There was no family history nor any other clinical signs of tuberous sclerosis.
The patient consented to undergo genetic testing, which was then performed on genomic DNA extracted from the peripheral blood by direct sequencing of the 17 exons of the BAP1 gene (MIM*603089). Primer pairs were designed according to the published reference genomic sequence (GenBank accession number NM_004656.3) to cover the coding sequence and the intron/exon regions of the gene. Analysis revealed the presence of a nonsense variant that was classified as likely pathogenic. After a few months, a compound melanocytic naevus on the patient’s back, with some dermoscopic atypias was excised. A BAP1-loss was equally demonstrated nevertheless it consisted of a pigmented macule, in contrast to the skin-coloured, dome-shaped lesions described previously.
Both the patient’s mother and brother underwent a total body skin examination. The mother was in her 60s and had Fitzpatrick’s phototype III with few naevi and no clinical or dermoscopic atypia. In contrast, the brother, a Fitzpatrick’s phototype II with multiple moles, had a skin colored, dome-shaped, slightly atypical naevus on his back with a dermoscopically spitzoid pattern that we decided to excise but the immunohistochemical study did not reveal loss of BAP1.Genotyping of the patient’s mother and brother revealed the presence of the same BAP1 mutation.
Fig. 1. Papule on the patient’s left hip. (a) Atypical ridge extending through a pink central dermal area with the presence of atypical peripheral polymorphic vessels at dermoscopic evaluation. (b) Histologically (haematoxylin and eosin (H&E), ×20), a compound naevus associated with an intradermal component of different-sized Spitzoid melanocytes surrounded by lymphocytes and without any mitoses. (c) Spitzoid cells negative for BAP1, and (d) positive for both p-16 and BRAF-v600E mutations.
BAP1 is a deubiquitinating enzyme, involved in various cellular processes such as cell cycle progression, cell differentiation and DNA damage responses (5). In melanocytes, which have a longer turnover than keratinocytes, BAP1-deficiency leads to an accumulation of DNA damage that increases the chance of neoplastic proliferation in patients with both sporadic and inherited deficiencies of BAP1 (6). BAP1 can achieve its tumour-suppressing role autonomously and in order to express the syndrome phenotypically, a double mutation (somatic and/or germline) is required.
BAP1-TPDS has been shown to follow an autosomal dominant pattern of inheritance and several different germline BAP1 mutations have been described, with more than 70% of them due to truncation of the gene (7). Our patient’s genetic test revealed the presence of a nonsense variant, c.1939G>T; p.Glu647Ter producing an early termination of the protein. This mutation variant has not been reported before in affected patients (8) and is not reported in the ExAc population database (9). Ultimately, the prevalence of this mutation in the general population is unknown; thus, the actual cancer risk in BAP1 carriers could be overestimated (10).
Although the complete phenotype of the syndrome is still being defined, there are certain cancer associations with BAP1 mutations, such as MMe, UM, RCC, CM, BIMTs, and BCC; the majority of them, except for MMe, seem to have a poor prognosis (2). Conflicting reports have been published regarding the association with several other cancers, such as breast carcinoma, cholangiocarcinoma, meningioma, neuroendocrine tumours, non-small cell lung adenocarcinoma and thyroid cancer (11). In addition to malignancies, patients carrying the mutation are more susceptible to develop many (5–50) skin lesions with polymorphic clinical appearances (12), frequently presenting as skin-colored, dome-shaped, well-circumscribed papules (13) with pink-to-tan structureless areas and peripheral irregular dots/globules or networks (3). BIMTs arise from conventional naevi, mostly with initiating oncogenic mutations in BRAF, which are represented by the lateral pigment globules visible on dermoscopy. A second subclone of larger epithelioid melanocytes, which usually lack pigmentation, could be determined by the inactivation of both alleles of BAP1; as a consequence, a central pink or flesh-colored, structureless area may be seen both clinically and dermoscopically (14).
The median age of onset of BIMTs in patients with germline BAP1 mutations is 32 (14).
It should be noted that BAP1 somatic inactivation is a frequent event in sporadic UM, MMe, and RCC; therefore, loss of BAP1 expression/LOH in an isolated UM, RCC, or MMe case with no family history of other tumours associated with the BAP1-TPDS is insufficient evidence for a pathogenic germline variant (1). This suggests that a family or personal history of cancer is necessary to justify genetic testing; however, there is still significant uncertainty in terms of the appropriate cancer surveillance warranted following a positive germline BAP1 mutation (10).
Star et al. (10) have proposed a surveillance model for germline BAP1 mutation carriers, which recommend the following: annual total-body skin examinations by dermatologists as well as dermoscopic follow-ups or excisions of BIMT-like lesions; yearly eye examinations and imaging by ocular oncologists; annual abdominal and respiratory clinical examinations with appropriate imaging (CT/MRI/US).
This case highlights the importance of skin evaluation in the early identification of a potential BIMT. In fact, in addition to a thorough and precise personal and family history, it could enable the appropriate referral of at-risk subjects to genetic counselling, with consequent cancer screening and pertinent follow-up.
We thank Antonio Percesepe (Medical Genetics Unit, Parma University Hospital, Parma, Italy) for his valuable comments.


 Click to show fullsize
Click to show fullsize