


1Department of Dermatology, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya, Aichi 466-8550, and 2Department of Dermatology, Fujita Health University School of Medicine, Toyoake, Aichi, Japan. *E-mail: makiyama@med.nagoya-u.ac.jp
Accepted Aug 6, 2019; E-published Aug 6, 2019
Oculodentodigital dysplasia (ODDD, OMIM#164200) is a rare, mostly autosomal dominant, congenital disorder with variable phenotypes. ODDD presents as craniofacial abnormalities, limb dysmorphisms, microdentia, and neurological and ocular abnormalities. ODDD is caused by a heterozygous mutation in GJA1 (OMIM#121014), which encodes the gap junction protein connexin 43 (Cx43). We report here a 2-year-old boy with ODDD whose main symptom at his initial visit was severe hypotrichosis.
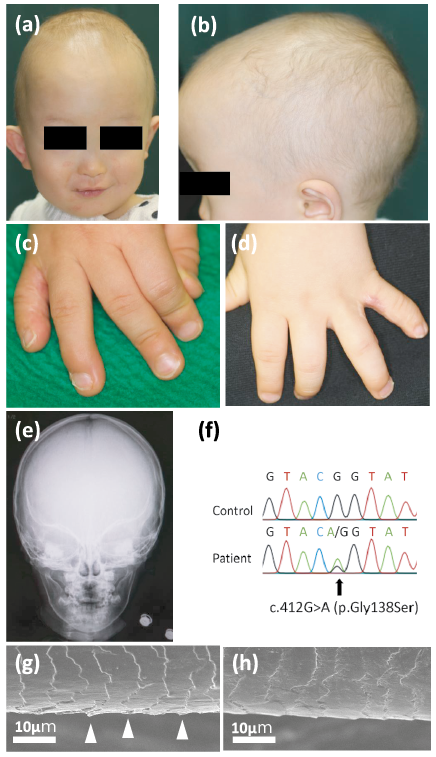
The proband is a 2-year-old Japanese boy who had presented with congenital alopecia. He showed severe hypotrichosis and low-set ears (Fig. 1a, b). Bilateral syndactyly of the 4th and 5th fingers was detected at birth (surgically repaired in infancy) and clinodactyly of the 5th fingers was also detected (syndactyly type III, OMIM#186100) (Fig. 1c, d). He had no craniofacial, ocular or neurological abnormalities, nor hypohidrosis, and a head X-ray was normal (Fig. 1e). He was born to non-consanguineous parents with no family history of any similar disorder. Ethical approval was obtained and all research was performed in accordance with the principles of the Declaration of Helsinki. Using genomic DNA as a template, genomic sequencing of GJA1 was performed as reported previously (1). A mutation search revealed a previously reported heterozygous missense mutation in GJA1, c.412G>A (p.Gly138Ser) (Fig. 1f). Neither of his parents had any mutation in GJA1. This suggested that the mutation was de novo. Scanning electron microscopy revealed weathering in the cuticle layer of the patient’s scalp hair (Fig. 1g), but not in the cuticle layer of age-matched control samples (Fig. 1h). Neither monilethrix nor hair nodules/beads were seen in the patient.
Fig. 1. Clinical features of the present patient. (a, b) The patient shows hypotrichosis of the scalp and low-set ears. (c, d) Bilateral syndactyly of the 4th and 5th fingers was surgically repaired in infancy, but clinodactyly of the 5th fingers remained. (e) No remarkable abnormalities are seen in the head X-ray. (f) Identification of the heterozygous missense mutation c.412G>A (p.Gly138Ser) in the GJA1 gene of the patient. The position of the mutation is indicated by the arrow. (g) Scanning electron microscopy reveals weathering (arrowheads) in the cuticle layer of the scalp hair, but not in (h) the cuticle layer of an age-matched control.
As a cause of ODDD, 3 mutations have been reported in the identical glycine residue at position 138: p.Gly138Ser, p.Gly138Arg and p.Gly138Asp. This suggests that the glycine residue in the cytoplasmic loop plays an important role in the function of Cx43.
In mouse models, 30% of p.Gly138Arg mutant mice are born with sparse hair, and the phenotype becomes more apparent in adulthood (2). In another study, approximately 20% of mice expressing p.Gly60Ser mutant Cx43 exhibited apparently lower hair density in the neck region. A histological comparison of the overall hair follicle density between p.Gly60Ser mutant mice and WT mice revealed no significant differences. However, after epilation or depilation, the mutant mouse hair was found to grow back slower, and hair growth was asynchronous in the mutant mice (3). These findings indicate that Cx43 is linked to a system that regulates the outward growth of the hair shaft. Cx43 has been reported to be expressed in the inner and outer root sheath, the dermal papilla, and the proliferating matrix in the rat hair follicle (4). In human skin, Cx43 is also reported to be expressed in keratinocytes, dermal fibroblasts, arrector pili muscles, sweat glands, sebaceous glands and hair follicles (5). We speculate that this linkage is involved in the pathogenesis of hypotrichosis from Cx43 mutations.
Indeed, in 26% of ODDD patients, poor hair growth or hair abnormality is observed (6). However, we were unable to find any report discussing the severity of hypo-trichosis in detail. Several reports showed photographs of the faces of patients, and their hypotrichosis was not severe in most cases (1, 6–8). We speculate that it is rare for patients with ODDD to show severe hypotrichosis.
Other than ODDD, there are many diseases that cause severe hypotrichosis. However, ODDD has several distinctive symptoms: craniofacial and limb dysmorphisms, spastic paraplegia and neurodegeneration. Craniofacial abnormalities (narrow and pinched nose with hypoplastic alae nasi, microdontia and enamel dysplasia) and limb abnormalities (syndactyly type III and bilateral syndactyly of the fourth and fifth fingers) are particularly important features for distinguishing ODDD from other diseases with hypotrichosis. In the present case, we suspected the diagnosis of ODDD from the syndactyly type III. The present case may suggest that we should suspect a diagnosis of ODDD when we see bilateral syndactyly even in a patient with severe hypotrichosis.
This work was supported by JSPS KAKENHI Grant Number JP18H02832 to M.A.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize