


1Center for Dermatology, Allergy and Dermatosurgery, HELIOS University Hospital Wuppertal, University Witten/Herdecke, Heusnerstr. 40, DE-42283 Wuppertal, 2Medical Clinic 1, Division of Oncology and Palliative Care, HELIOS University Hospital Wuppertal, Wuppertal, 3Department of Dermatology, University of Cologne, 4Department I of Internal Medicine, Center for Integrated Oncology Aachen-Bonn-Cologne-Duesseldorf, Excellence Cluster for Cellular Stress Response in Aging-Associated Diseases, and Center for Molecular Medicine Cologne, University of Cologne, Cologne, and 5University of Witten/Herdecke, Germany. E-mail: silke.hofmann@helios-gesundheit.de
Accepted Aug 12, 2019; E-published Aug 13, 2019
T-cell prolymphocytic leukaemia (T-PLL) is a rare and aggressive haematological malignancy that typically presents with hyperlymphocytosis, splenomegaly and lymphadenopathy, but also with frequent cutaneous involvement (1, 2). To raise awareness of potential cutaneous presentations underlying a T-PLL, we report here 2 patients who both initially had therapy-resistant pruritus and subsequently developed progressive cutaneous lesions mimicking atopic eczema.
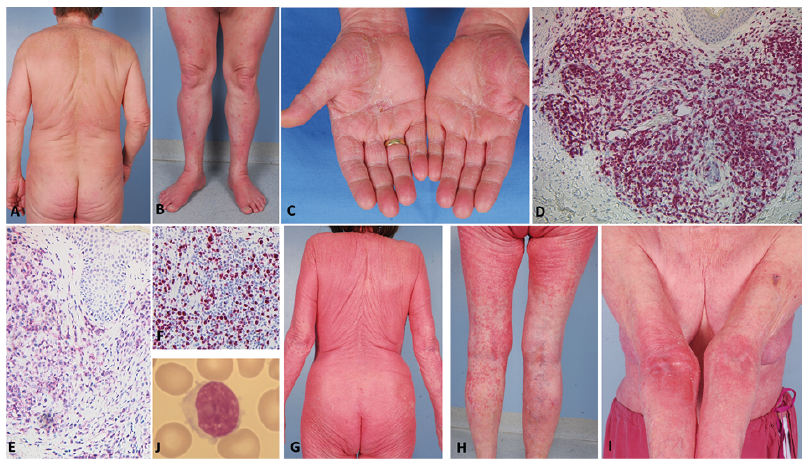
Patient 1. A 62-year-old man with a history of type 2 diabetes mellitus and depression initially presented with therapy-resistant generalized pruritus for the past 3 years. Examination revealed a very small number of erythematous, excoriated small papules on his trunk and legs (Fig. 1A, B), xerosis cutis, Herthoge’s sign positivity, Dennie-Morgan infraorbital folds, and a white dermographism. Laboratory parameters showed a mild peripheral blood (PB) leukocytosis (12/nl, normal < 10) with a lymphocytosis of 4.68/nl (< 3.0), eosinophilia of 6% (< 4), and a serum lactate dehydrogenase (LDH) of 421 U/l (< 266). Liver and kidney tests were within normal limits, as were total IgE, C-reactive protein (CRP), thyroid stimulating hormone (TSH), vitamin, and ferritin levels. Histology of an erythematous papule demonstrated spongiotic dermatitis.
In the following months, the patient was hospitalized repeatedly due to persistent pruritus and newly developed palmar keratoderma (Fig. 1C). He received treatments with corticosteroids, photo-therapy, and H1-blockers, all without significant benefit. Five months after initial presentation the patient reported weight loss of 7 kg within 2 months. Clinical examination revealed hepatosplenomegaly and generalized lymphadenopathy. A mild leukocytosis (10.18/nl) entailed a lymphocytosis of 5.13/nl. Cytomorphology of PB smears revealed prolymphocytes with convoluted nuclei. The lymphocytes were predominantly mature CD4+ T cells, which expressed CD2, CD5 and CD7. Refined flow cytometry showed intracellular expression of the TCL1A oncogene and a clonal dominance of the TCR-Vß-1 chain. Fluorescence in situ hybridization (FISH) analysis detected a deletion of TP53, a MYC amplification, and a 14q11-rearrangement. Those findings led to the diagnosis of mature T-cell leukaemia with cutaneous tropism, precisely T-PLL. A skin biopsy showed dense perivascular dermal infiltrates with atypical lymphocytes expressing CD3 and CD4 (Fig. 1D–F), but no epidermotropism. The patient received alemtuzumab therapy (anti-CD52 antibody) and his pruritic skin lesions resolved rapidly, but cessation of antibody therapy was necessary due to reactivation of cytomegalovirus. Six months after initial diagnosis an allogeneic stem cell transplantation (ASCT) was performed due to progressive disease. The subsequent complete remission is ongoing since August 2015.
Fig. 1. Clinical and immuno-histochemical findings. (A) Minimal eczema on the back and (B) pruritic papules on the lower legs (B) at initial presentation of patient 1. Two months later, he presented with additional palmar hyperkeratosis (C). Immunohistochemistry of skin lesions showed expression of (D) CD3 and (E) CD4 within dermal lymphocytic infiltrates (x200). (F) Proliferation marker Ki67 was expressed in 70% of the lymphocytes. (G-I) Patient 2 presented with generalized maculopapular erythematous lesions with exfoliation and erythematous infiltrates in the arm folds one year prior the diagnosis of T-PLL. (J) A blood smear showed medium sized lymphatic cells with oval-shaped nuclei, intermediate chromatin density and prominent eccentric nucleoli suggestive of prolymphocytes.
Patient 2. An 83-year-old woman presented in 2015 with a 10-year-history of extremely pruritic generalized maculopapular exfoliative rashes refractory to topical corticosteroids and ultraviolet A (UVA)-phototherapy (Fig. 1G–I). Histology showed superficial spongiotic dermatitis and total IgE was elevated (214 kU/l, normal < 100) leading to the diagnosis of late-onset atopic eczema. A leukocytosis of 14.83/nl was noted at initial presentation, but was attributed to previous systemic corticosteroid therapy. Treatment with cyclosporine A (CSA) 200 mg/day in addition to topical potent corticosteroids was introduced.
The patient was re-admitted one year later for surgical treatment of newly developed Bowen’s disease and actinic keratoses on the face. The rash, however, had improved markedly since the introduction of CSA, but on clinical examination, axillar and inguinal lymphadenopathy was noted. Blood tests revealed persistent leukocytosis (17.34/nl), lymphocytosis (10.41/nl), low eosinophils (0.3/nl) and increased LDH (355 U/l). The PB revealed 10/nl lymphoma cells with a classical prolymphocytic appearance (smear in Fig. 1J) showing medium-sized lymphatic cells with oval-shaped nuclei, intermediate chromatin density and prominent eccentric nucleoli. Repeated nuclear irregularities, but no cerebriform appearance, were present. Flow cytometry demonstrated in the lymphocyte gate 97% T cells (CD4/CD8 ratio of 96), 1% B cells, and 1% NK cells. All 4 pan-T-cell antigens were expressed without significant losses: s-CD3 on 98%, CD2 on 98%, CD7 on 95%, and CD5 on 89% of lymphocytes. This raised the suspicion of T-PLL, but further confirmatory tests were not performed as the patient refused further examination and more intense treatments. Within the next 2 years, she developed symmetrical palmoplantar hyperkeratosis, but was in good general condition. In August 2018, the patient reported tiredness, and leucocytosis had increased to 29.1/nl. Given her age, she preferred continuation of CSA and topical corticosteroids without further tests.
We report here 2 patients presenting with refractory long-standing pruritus alongside mild-to-severe eczematous manifestations initially misdiagnosed as atopic eczema. In both, a diagnosis of T-PLL had eventually been established. The first patient was eligible for an ASCT, which finally conferred long-term disease control. Both cases instructively demonstrate that a prodromal period of cutaneous symptoms can exist in T-PLL. In fact, from coincidental diagnoses of T-PLL, it is known that there is an indolent early phase in approximately 20–30% of cases, in which the initially mild lymphocytosis goes unnoticed. The prominent pruritus in the patients reported here most likely led to the detection of T-PLL at such an early phase.
In general, T-PLL is a rare leukaemia typically occurring in the 7th decade of life. T-PLL usually shows PB lympho-cyte doubling times of < 6 months with bone marrow infiltration, hepatosplenomegaly, lymphadenopathy, and follows an aggressive clinical course with median survival times of < 2–3 years (3, 4). The T-PLL cell is a mostly CD4+ post-thymic T cell with strong expression of CD7. Cytogenetics characteristically reveals expression of the oncogene TCL1A.
Skin involvement has been described in T-PLL and requires informed differentiation from primarily cutaneous T-cell neoplasms, such as Sézary syndrome (2, 4, 5). To further evaluate the reported cases in the context of the incidence and spectrum of cutaneous manifestations in T-PLL, we retrospectively analysed clinical data of 107 patients with T-PLL from our second-opinion haematology clinic. Overall, we detected 29 patients (27.1%) who presented with cutaneous involvement, consistent with published data (2, 4, 5). The pattern of T-PLL skin lesions was very heterogeneous, including maculopapular rashes, indurated lesions, periorbital oedema, conjunc-tival infiltration, purpura, palmoplantar hyperkeratosis, and erythroderma. Other studies report similar lesions, additionally describing patients with nodules, vesicles, erosions or exfoliative dermatitis (6, 7).
In our “haematology” cohort, facial skin and/or conjunc-tivae were involved in 13/107 cases and trunk and limbs in 12 and 9 patients, respectively. Magro et al. reported facial involvement with purpura or oedema in 5 of 6 (2). The 2 patients reported here did not show predominant facial lesions. Instead, they had extremely pruritic eczematous skin manifestations. Pruritus may be associated with neoplasms of the hepatobiliary tract, the skin, and the hematopoietic system (8). In cutaneous T-cell lymphoma, 88% of patients have intractable (non-histaminergic) pruritus mediated by increased expression of interleukin 31 (9). Furthermore, both patients developed palmoplantar hyperkeratosis. This phenomenon, as well as severe pruritus, has been described in T-PLL, but is also a characteristic finding in patients with Sézary syndrome. The latter can be differentiated from T-PLL by its frequent erythrodermic manifestation, gyrated cerebriform nuclei, and a lack of expression of CD7 (4).
In conclusion, accurate evaluation of non-specific pruritic skin lesions associated with haematological abnormalities can lead to the early detection of T-PLL. Avoiding a protracted diagnosis and early engagement in an interdisciplinary approach may allow for better outcomes of this otherwise aggressive malignancy.


 Click to show fullsize
Click to show fullsize