


1Department of Dermatology, University Hospital Zurich, Gloriastrasse 31, CH-8091 Zürich, 2University of Zürich, Zürich, 3Department of Dermatology and Allergology, and 5Department of Gynecology and Obstretics, Cantonal Hospital St Gallen, St Gallen, Switzerland, 4AllergyCare Allergy Diagnosis and Study Center, Vienna, Austria, 6Department of Dermatology, University Medical Center Freiburg, Faculty of Medicine, Freiburg, Germany, 7Human Genetics Laboratory Genetica, Zürich, Switzerland, 8Department of Dermatology, Eberhard Karls University, Tubingen, Germany, 9Department of Pediatric Dermatology, University Children’s Hospital Zurich, Switzerland, 10Department of Dermatology and Allergy, University Hospital, LMU Munich, Munich, Germany, and 11University of Lausanne, Switzerland. E-mail: emmanuella.guenova@gmail.com
#These authors shared last authorship.
Accepted Sep 10, 2019; Epub ahead of print Sep 10, 2019
Acta Derm Venereol 2020; 100: adv00041
Epidermolysis bullosa (EB) is a group of genetic disorders characterized by fragility of skin and mucosa. The majority of subtypes of EB result from malfunction of anchor proteins of the basal membrane zone (BMZ). Based on the level of blister formation, EB is classified into 4 major forms: EB simplex, junctional EB, dystrophic EB, and Kindler syndrome (Fig. S1).
Junctional EB (JEB) is caused by mutations in genes encoding for components of the lamina lucida, e.g. integrin beta 4 (ITGB4), integrin alpha 6 (ITGA6), laminin-332 genes (LAMA3, LAMB3 and LAMC2), etc. One specific type of JEB with mutations in α6β4 integrin presents with skin fragility in association with pyloric atresia. JEB is inherited only in a recessive manner.
Trisomy 2 mostly results in first trimester pregnancy loss; it is compatible with life exclusively in mosaic states or when restricted to the placental tissues. Common in utero indications of mosaic trisomy 2 are oligohydramnios and poor foetal growth. High trisomic levels in placenta and the foetus can result in abnormal and fatal phenotypes (1). Phenotypes of uniparental disomy of chromosome 2 (UPD2) resulting in trisomy mosaicism have described phenotypes, such as genital hypoplasia, intrauterinal growth retardation (IUGR) and oligohydramnios. Exceptional cases of full UPD2 have been described in healthy individuals (2, 3).
We report here a case of a newborn with JEB resulting from a previously unknown homozygous mutation leading to a premature termination codon in the gene encoding for ITGA6. The loss of heterozygosity occurred in the setting of an exclusive maternal UPD2.
A 28-year-old healthy primigravida underwent amniocentesis after ultrasonography performed at 31 weeks of gestation revealed foetal abnormalities (growth retardation, pyloric atresia with polyhydramnios and a fluid-filled stomach, and complex lower-limb anomalies with disproportional long-bone shortening and aplasia of the foot structures).
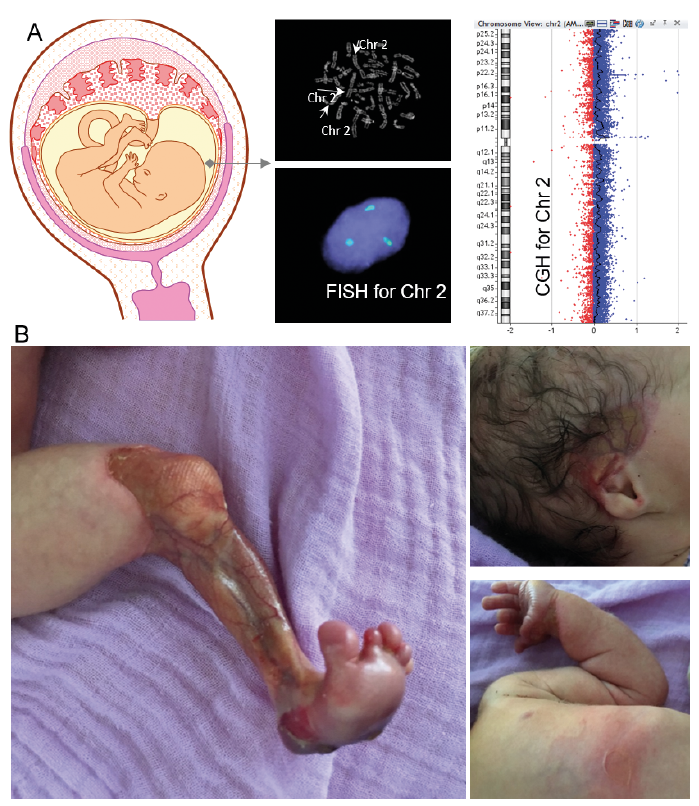
Amniotic fluid was submitted for cytogenetic analysis and array-based comparative genomic hybridization (CGH) testing. Chromosomal analysis and metaphase fluorescence in situ hybridization (FISH) for chromosome 2 revealed a rare trisomy 2 in 12–15% of all cells in the amniotic fluid with an aberrant 47, XX +2 (2)/ 46, XX karyotype, confirmed by array-based CGH (Fig. 1A).
Due to foetal growth arrest and polyhydramnios, labour was induced at 37 weeks’ gestation. The newborn was small for gestational age (3 standard deviations (SD) below 1st percentile). Multiple blisters of the skin on the trunk, head, and umbilical cord could be observed, as well as aplasia cutis on all 4 limbs (Fig. 1B). The infant died after 5 days.
Fig. 1. Prenatal and clinical findings. (A) Amniocentesis results at 31 weeks’ gestation. Chromosomal analysis with karyotyping and metaphase fluorescence in situ hybridization (FISH) for chromosome 2 shows trisomy 2 in 12–15% of all cells in the amniotic fluid. Aberrant 47, XX +2 (2)/ 46, XX karyotype. The comparative genomic hybridization (CGH) array distribution indicates a higher amount of Chr 2 in the patient than in the reference DNA. (B) Clinical images of the patient 1 day after birth with skin aplasia of the left leg, blister on the right side of the head and ear and new blisters following minor trauma (touch) on the torso and left arm.
Histology revealed a subepidermal blister without inflammation. The basal membrane, visualzed by staining for collagen type IV, remained firmly attached to the base of the blister cavity. The ultrastructural sites of the blister formation could be detected within the lamina lucida. There was a decrease in the transmembrane proteins of the hemidesmosomal plate. Immunofluorescence mapping disclosed junctional splitting and loss of immunoreactivity for the integrin a6/ß4 (Fig. S2A–E).
Whole exome sequencing (WES) ruled out non-paternity and identified a novel frame-shift mutation in the ITGA6 gene for integrin α6 (patient homozygous, mother heterozygous, and mutation absent from the father) and a missense variant in the COL4A4 gene (patient homozygous, mother heterozygous, and mutation absent from the father). Collagen type IV protein was unaffected both immunohistochemically (Fig. S2B) and on immunofluorescence mapping (Fig. S2E); however, the high pathogenicity of the mutation indicates a possible clinical relevance. COL4A4 mutations have not previously been described in any EB subtype.
The mutation in ITGA6 has not been reported previously. The CADD Phred score of 35 for the ITGA6 mutation places it in the top 0.1% of altering mutations. This novel frame-shift mutation in the ITGA6 gene accounted for the lack of integrina6 protein in the patient’s skin and was the pivotal feature to diagnose an autosomal recessive inherited junctional epidermolysis bullosa with pyloric atresia (JEB-PA). In confirmation of the results of the immunofluorescence mapping (Fig. S2E) and the WES, Sanger sequencing disclosed a homozygous insertion c.2843_2844dupCG, in exon 22 of the JEB-associated gene ITGA6 (chr2exon22 c.2843_2844dupCG) (Fig. S3A–B). The 2-nucleotide insertion resulted in a frame-shift mutation and a premature stop codon and termination of translation p.Ser949Argfs*15. The mutation was located on chromosome 2, an extra copy of which (trisomy 2) we had identified previously in 12–15% of all cells in the amniotic fluid during the prenatal diagnostic at 31 weeks’ gestation.
At birth, the loss of evidence for persistent trisomy 2, and the clinical manifestation of an autosomal recessive disease in a child born to a wild-type father and a heterozygous mother, suggests a complete chromosomic rescue and unipaternal (maternal or paternal only) inheritance of the genes located on the affected chromosome 2.
For an accurate evaluation of the suspected UPD2, postnatal segregation analysis with chromosome 2-specific short tandem repeat (STR) markers was performed. This exhibited exclusive maternal and a lack of any paternal inheritance of chromosome 2 in the newborn, the maternal uniparental isodisomy 2, following the complete chromosomic rescue of trisomy 2, unmasked in a homozygous setting the novel chr2exon22 c.2843_2844dupCG mutation in the ITAG6 gene and resulted in the clinical manifestation of a lethal ITGA6-related JEB-PA.
Foetal trisomy can occur due to meiotic non-disjunction events in either parent (4). Trisomy rescue is a rare phenomenon whereby the zygote, with 3 copies of a chromosome, spontaneously loses 1 copy, resulting in a diploid state. Indeed, any copy of the chromosome can be lost, potentially retaining both copies from the faulty gamete, resulting in a UPD (Fig. S4), identical alleles (isodisomy) of multiple genes, and may be the cause of the clinical manifestation of imprinting or recessive disorders. The rate of segmental UPD is estimated as 1 per 3806 chromosome pairs (0.026%) (5). Whole-chromosome UPD is very rare.
A few cases of JEB have been described with a UPD-based inheritance pattern. Mutations in laminin-332 are associated with the severe subtype of severe generalized (Herlitz) JEB (6). Homozygous mutations in 2 subunits of laminin 332 (LAMB3 and LAMC2) caused by maternal and paternal UPD of chromosome 1 have been reported a few times (7–11). Inheritance of JEB with pyloric atresia through a UPD event has been described (12).
ITGA6 is an important component in embryogenesis with roles in endoderm migration and nervous system development. It forms a heterodimer with ITGB4 (α6β4) in the BMZ. In JEB the ultrastructure of hemidesmosomes is abnormal, and several mutations in ITGA6 and ITGB4 have been reported (13). Some mutations result in less severe phenotypes with longer survival rates; hence the characterization of all ITGA6 and ITGB4 mutations is important to assist patient counselling (14). In the case reported here, the newly detected chr2exon22 c.2843_2844dupCG mutation is strongly indicative of a severe phenotype with a short survival.
This project was supported by the Jubiläumsstiftung von SwissLife, the Promedica Stiftung (1406/M and 1412/M), a further anonymous Foundation, the Swiss Cancer Research Foundation (KFS-4243-08-2017) and the Clinical Research Priority Program (CRPP) of the University of Zurich.


 Click to show fullsize
Click to show fullsize