


1First Department of Pediatrics, Medical School and 4First Department of Pediatrics, Neurology unit, Medical School, National and Kapodistrian University of Athens, “Agia Sofia Childrens Hospital”, Athens, Greece, 2Neonatal Intensive Care Unit and 3Department of Pediatric Surgery, General Hospital of Nikaia “Agios Panteleimon”, Peiraius, Greece, 5Department of Dermatology, Faculty of Medicine, Medical Center, University of Freiburg, Hauptstr. 7, DE-79104 Freiburg, and 6Department of Medical Genetics, Institute of Mother and Child, Warsaw, Poland. *E-mail: dimitra.kiritsi@uniklinik-freiburg.de
Accepted Sep 12, 2019; E-published Sep 12, 2019
Epidermolysis bullosa (EB) is a clinically and genetically relatively heterogeneous group of inherited blistering disorders. The most common subtype is EB simplex (EBS), which is usually associated with mutations in the KRT5, KRT14, DST, EXPH5 and PLEC genes (1). Approximately 8% of patients with EBS are estimated to carry PLEC mutations (2). The PLEC gene encodes the large cytolinker protein plectin (3–5). Plectin mutations, inherited in an autosomal recessive pattern, result in distinct phenotypes, including EBS with muscular dystrophy (MD), EBS with pyloric atresia (PA) and EBS with skin lesions only (3, 6). The dominantly inherited EBS-Ogna has a mild course restricted to skin involvement (7).
Phenotype–genotype correlations have suggested that EBS-MD is mostly due to genetic variants in the central rod domain of plectin and EBS-PA due to mutations outside this domain (3, 8). Urinary tract involvement has only very rarely been reported in patients with either EBS-PA or EBS-MD (3, 4). To our knowledge only one case of EBS with both PA and MD, due to compound heterozygous PLEC mutations located in exon 32, has been reported (9). That patient had severe skin involvement and died 3 months after birth. We report here a patient with non-lethal EBS with rather mild skin involvement, congenital PA, progressive MD and mild bilateral hydronephrosis, who is compound heterozygous for 2 PLEC genetic variants in exon 32, one of them not having been reported previously. Thus, we extend the phenotypic spectrum of genetic PLEC variants.
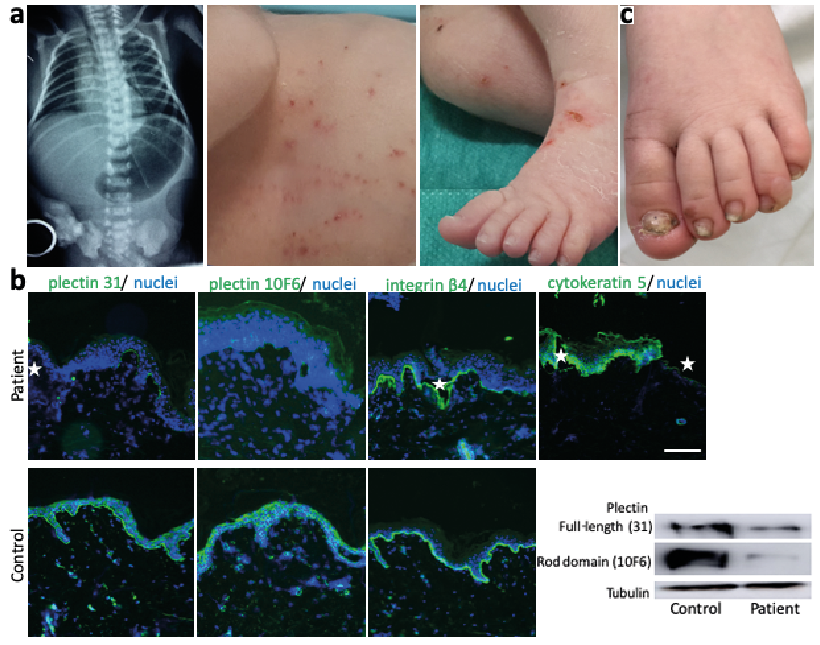
On the second day of life, an infant boy was observed to have non-bilious vomiting with blood, without abdominal distention. He had been born at 38 weeks’ gestation, with a weight of 2,400 g. Abdominal X-ray showed a large gastric air bubble with no gas distally (Fig. 1A). An upper gastrointestinal tract examination with contrast material revealed a dilated stomach with obstruction at the pylorus, consistent with PA. Tense, and often haemorrhagic, blisters and erosions on the trunk and extremities appeared and recurred at sites of mechanical trauma (Fig. 1A). Similar bullous lesions were noted in the oral mucosa. Creatine kinase (CK) was found 1,468 U/l (normal range < 350 U/l) on the 5th day of life. On the 6th day of life, laparotomy revealed PA type 2 (pyloric canal replaced by a solid cord of tissue) and gastroduodenostomy was performed. Feeding was started approximately 1 week after surgery, was well tolerated, and hence gradually increased to full enteral nutrition. A renal ultrasound at day 8 showed mild hydronephrosis. Skin lesions continued to recur, and healing occurred without scarring, accompanied by mild atrophy. A skin biopsy was taken for immunofluorescence mapping, using a panel of antibodies to components of the dermo–epidermal junction and showed intraepidermal blisters and strongly reduced staining with 2 antibodies against plectin (Fig. 1B, 2 left panels). All other markers stained similarly to control skin. These findings were consistent with the diagnosis of EBS due to PLEC mutations. A custom-designed panel of EB-related genes was analysed by the Department of Medical Genetics in Warsaw, Poland, using next generation sequencing. The patient is compound heterozygous for the novel mutation c.11912del, p.Lys3971ArgfsTer10 and the mutation c.12499C>T, p.Arg4167Ter, both located in exon 32 of the PLEC gene (name according to coding ref seq: NM_000445.3). Immunoblot with antibodies recognizing the C-terminus and the rod domain of plectin both revealed strongly reduced levels of apparently full-length plectin (Fig. 1B, right panel), although a slightly smaller, truncated form cannot be excluded. All analyses were performed after ethics approval and informed consent, and in accordance with principles of the Declaration of Helsinki.
At the age of 5 months the patient had kept up with his main developmental milestones. One month later, CK was evaluated while suffering from an acute viral respiratory illness with hypotonia and was greatly increased at 28,068 U/l. Follow-up CK levels were obtained at the age of 14 months and 17 months and were slightly above the upper range, at 184 U/l and 206 U/l, respectively (normal range <140 U/l). He had growth and motor developmental delay and only at approximately the age of 20 months could he walk and stand without help. He continued to develop sparse blisters, mostly on the extremities, and had nail involvement with thickened dystrophic nails on fingers and toes (Fig. 1C). Hydronephrosis had remained stable, without signs of renal parenchymal disease. No cardiomyopathy was detected during all this time.
Fig. 1. Clinical picture of the patient and diagnostics compatible with the diagnosis of epidermolysis bullosa simpelx due to PLEC mutations. (a) Abdominal X-ray after birth showed a large gastric air bubble with no gas distally. Small blisters and erosions were present over the whole integument. (b) Immunofluorescence mapping reveals intraepidermal blisters (white stars in plectin, integrin β4 (clone 3Ε1) and cytokeratin 5 (clone D5/16 B4) staining) and strongly reduced plectin expression with 2 different antibodies, whereas integrin a6 shows a staining pattern comparable to control skin. Scale: 100?μm. Immunoblot with the 2 plectin antibodies (31 recognizing the C-terminus and 10F6 for the rod domain) reveals residual presence of full-length plectin in lysates from patient keratinocytes, although a slightly shorter fragment cannot be fully excluded. Plectin expression appears to be comparably strongly reduced, regardless of the epitope recognized by the 2 plectin antibodies. (c) At the age of 2 years the skin phenotype has improved with mostly acral blisters and thickened dystrophic nails.
Plectin is a protein involved in cytoskeletal organization (8, 10). It comprises N- and C-terminal domains, which contain multiple protein–protein interaction sites, separated by an elongated central rod domain. The human PLEC has 8 isoforms formed by alternative splicing of exon 1. In normal tissues both full-length and rodless plectin is expressed. The rodless isoforms are produced through alternative splicing of exon 31, which encodes the rod domain. Patients with EBS-MD have been described to lack full-length plectin with the maintenance of the rodless isoform. The patients with EBS-PA described so far had PLEC mutations outside of exon 31, resulting in loss of both full-length and rodless plectin and thus associated with early lethality (8). A few EBS-PA cases survived longer after surgery; one has been reported at the age of 6 months (11), and another at the age of 12 years (3). These patients showed mild cutaneous phenotypes. Development of MD in a case with PA has not been reported so far, except for one patient (9) who was compound heterozygous for PLEC mutations located in exon 32. Another case of a 6-year-old girl with EBS-PA additionally experienced significant urological abnormalities and showed slightly increased levels of CK. Genetic analysis disclosed 2 premature stop codon mutations in exons 23 and 31 of PLEC (4).
The patient described here is, to our knowledge, the second case of EBS, with PA and MD due to PLEC mutations, and the only one who is alive and thriving at the age of 2.5 years. Patients with EBS-MD usually have mild skin bullous lesions and nail dystrophy, as in our patient. It remains to be established whether patients with EBS-PA would also develop MD that might have been missed in other cases, since almost all reported patients with EBS-PA died during the first months of life, and the MD is initially asymptomatic and slowly progressing. Our patient had elevated CK levels on the 5th day of life, a screening marker in a variety of congenital muscular dystrophies (12). The connection of acute illness-associated weakness with high levels of CK (>10× normal) and then lower, but persistent elevation of CK has been described in young patients with congenital muscular dystrophies (13). A muscle ultrasound of the patient at the age of 2.5 years of the anterior forearm and upper extremity did not detect increased echo intensity and thus was reported as normal (14). The presence of strongly reduced, but apparently full-length, plectin with immunoblot using antibodies recognizing the C-terminus and the rod domain explains the non-lethal phenotype in our patient.
The authors thank the patient and his family. We greatly acknowledge the excellent technical assistance of Kaethe Thoma and Annegret Bedorf.


 Click to show fullsize
Click to show fullsize