


Departments of 1Dermatology, University Hospital of Münster, Von-Esmarch-Straße 58, DE-48149 Münster, 2Division of Pediatric Stem Cell Transplantation and Immunology, 3Department of Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, 4Center for Pediatrics and Adolescent Medicine, 5Institute for Immunodeficiency, Center for Chronic Immunodeficiency (CCI), Medical Center - Faculty of Medicine, University of Freiburg, Freiburg, and 6Department of Pediatrics, University Hospital of Münster, Münster, Germany. E-mail: Kira.Suessmuth@ukmuenster.de
Accepted Sep 25, 2019; E-published Sep 25, 2019
Acta Derm Venereol 2020; 100: adv00002.
Prolidase deficiency (PD) (OMIM170100) is a rare genetic metabolic autosomal recessive disorder caused by mutations in the PEPD gene encoding the enzyme prolidase D. Prolidase divides peptides containing proline and hydroxyproline (1, 2) and plays an important role in the catabolism of collagens (3).
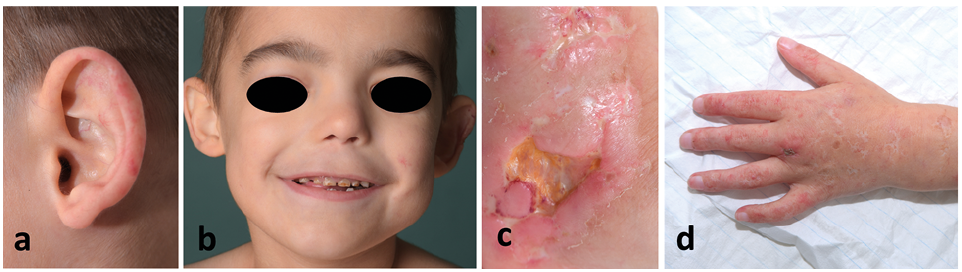
The resulting phenotype is diverse: recurrent infections – especially of the respiratory tract – teleangiectasia similar to the phenotype we know from collagenoses, dysmorphic facial findings (Fig. 1), hepatosplenomegaly, delay of development and in many cases painful ulcerations. Thrombocytopenia, hypocomplementemia and hypergammaglobulinemia are frequent laboratory findings (4) with imidodipeptiduria (10–30 mmol/day) as the typical finding in urine amino acid analysis (5).
The therapy of PD remains challenging. All therapies described are symptomatic ones. There is only insufficient data on pain and wound management. Recently, hyperbaric oxygen therapy has been described as a possible treatment for ulcers that reduces exudation and supports granulation (6).
Fig. 1. Phenotype of patient with prolidase deficiency. Phenotype of prolidase deficiency includes teleangiectasia (a), hypertelorism and a flat bridge of the nose (b). Note deep ulceration (c) and scarring (d). Permission is given to publish these photos.
We present a 6-year old boy diagnosed with PD consulting a dermatological clinic because of painful, chronic wounds on the extremities, especially on the feet. The patient shows a homozygous mutation in the PEPD gene (mutation c.1342G>A;p.Gly448Arg) detected by exome sequencing after urine analysis had revealed massive imidodipeptiduria.
The parents (both heterozygous mutation carriers) are not related and have a healthy older daughter. The first symptoms referring to a severe systemic disorder appearing soon after birth were thrombocytopenia, hypochromic microcytic anaemia, hepatosplenomegaly, Iymphadenopathy and numerous infections. Later he developed severe diarrhoea that has been partly controlled under therapy with steroids. Differential diagnoses considered initially were neonatal lupus erythematodes (NLE), hemophagocytic Iymphohistiocytosis (HLH), and autoimmune Iymphoproliferative syndrome (ALPS).
The first skin lesions appeared approximately 3 weeks after birth with papules and livid spots turning into lesions resembling livido racemosa covering the extremities and face. Later these lesions ulcerated. The histopathology taken from the intact periphery of one of the ulcers revealed multiple thrombotic cutaneous vessels with fibrinoid degeneration of the vessel walls. Apart from some extravasated erythrocytes, inflammatory cells were almost absent. The epidermis was hyperplastic and showed signs of degeneration and paraceratotic cornification. The dermal connective tissue appeared fibrotic (Fig. S1). The tissues taken from the gastrointestinal tract showed similarities to Crohn’s disease.
Apart from antibiotics, the patient was treated with immuno-suppressive drugs assuming HLH and ALPS. Later he received rituximab as an immunomodulatory therapy which caused an immunoglobulin deficiency.
We saw the boy for the first time with multiple painful, necrotic ulcers and one ischaemic toe (Fig. S2a–d). He was not able to walk anymore due to pain and was treated with oral antibiotics, low dose prednisolone, steroid pulses and sirolimus for more than 5 years. Wound therapy was performed with standard wound procedure. His ulcers showed similar symptoms, clinical configurations and histopathological features to those found in livedoid vasculopathy (LV). Therefore, we started a therapy with low-molecular-weight heparin once a day (1 mg/kg body weight) and documented follow-ups 5 weeks, 22 weeks and 13 months after the first injection.
During the period of treatment with anticoagulation, the therapy with steroids was continued because of diarrhoea. Sirolimus was stopped when low dose prednisolone was taken continuously approximately 3 months after the first visit to our clinic. Eight months after the first heparin injection, the patient also started a therapy with a TNF-α-inhibitor (adalimumab) as a steroid sparing agent due to ongoing gastrointestinal symptoms.
Ischaemia disappeared 5 weeks after the first injection (Fig. S2f) and there was a slight improvement of the pain situation (from 9 on the numeric rating scale (NRS) to 7). At the third visit, even fibrin clearly disappeared and the small wounds healed (Fig. S2i–l). The therapy with heparin has not been interrupted. After the fourth visit, we observed an almost complete healing and the patient had no pain (NRS 0). He started to relearn walking.
Adalimumab did not show a long-term effect on the gastrointestinal symptoms. Furthermore, the boy developed a severe respiratory infection as a possible side effect of immunosuppression. Injections with adalimumab were stopped. Wound conditions stayed stable without any new ulcerations under treatment with anticoagulation.
Our case report shows that PD can cause thrombosis in the cutaneous microcirculation similar to observations in LV. The cause for vasculopathy remains unclear. Analysis of coagulation and autoimmune parameters revealed elevated levels of fibrinogen, IL-2, IL-6, and ferritin due to inflammatory processes which might explain thrombophilia. Analysis of activated partial thromboplastin time (aPTT) and standard coagulation parameters did not reveal any pathological findings. aPTT twice showed increased values without any clinical correlation. The values quickly normalized over time without any treatment. Thrombocytes were always low (between 150 and 250 tsd./μl) as typical for PD. In literature, some further aspects are discussed to cause necrosis: existence of antinuclear antibodies or hypergammaglobulinaemia (7, 8) and an accumulation of antibodies or dipeptides in the small vessels due to a missing prolidase activity (9). Kryoglobulines could histologically be excluded in our patient. Moreover, secondary thromboembolism due to damage in the small vessels because of a disturbed collagen metabolism might explain vasculopathy.
Apart from vasculopathy as the main pathomechanism, the skin seems to be fragile in general. Therefore, we assume further pathomechanisms that explain prolonged wound healing: missing trace elements (decreased zinc levels in our patient) due to massive diarrhoea seem to play an important role, too.
The influence of other medication on wound healing can be discussed, but the only newly initiated substance during treatment with heparin was a TNF-α-inhibitor after 8 months of anticoagulation. Before starting this new therapy, we had already observed a clear tendency of healing (Fig. S2a–l) and there was no rebound after stopping the injections. In this single case report, our observation might be a coincidence. However, in this patient ulcerations were thus far a lifelong existing symptom that rapidly showed a stable improvement after the beginning of anticoagulation. Moreover, we can show that vasculopathy was histologically present in this patient. To further underline our findings, more patients with PD and vasculopathy need to be observed.
A reduction of the heparin dose or changing to an oral anticoagulation could be another option for our patient (10, 11), but there might be a specific benefit of using enoxaparin in PD. Prolidase directly binds coagulation factors. This mechanism leads to a degradation of prolidase. Recently it has been shown that enoxaparin elevates plasma levels of prolidase in mice by inhibiting this proteolysis pathway. It is assumed that this pathway is involved in inflammatory processes, immune response and vascular physiology. However, further studies of patients with PD are necessary to understand how altered or missing prolidase influences this pathway (12).
Our case report leads to a more precise understanding of the pathomechanism of the skin lesions in PD and opens the avenue for novel therapeutic approaches.
We thank the family of our patient for their cooperation and continuous exchange. We also gratefully thank all colleagues, especially those involved in pediatrics, who closely cooperated with us.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize