


Department of Dermatology, Faculty of Medicine and Graduate School of Medicine, Hokkaido University, North 15 West 7, Kita-ku, Sapporo 060-8638, Japan. *E-mail: nomura@huhp.hokudai.ac.jp
Accepted Sep 25, 2019; E-published Sep 25, 2019
Nagashima-type palmoplantar keratosis (NPPK), the most common palmoplantar keratoderma (PPK) in East Asia, is characterized by well-demarcated erythema with mild-to-moderate hyperkeratosis on the palms and soles with transgrediens (1, 2). Biallelic loss-of-function mutations in SERPINB7 are responsible for NPPK (1, 2). We report here a case of NPPK with malignant melanoma (MM).
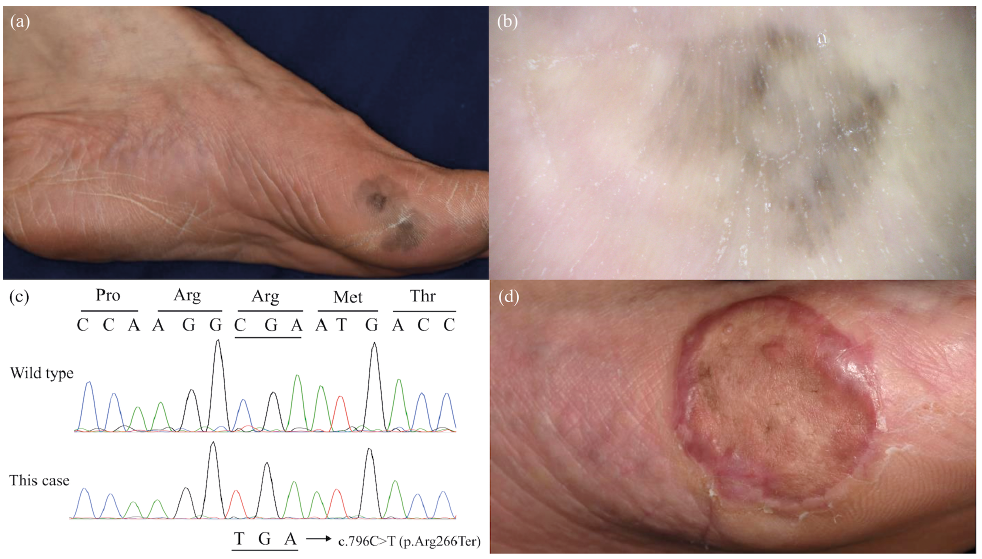
A 45-year-old Chinese woman presented to our hospital with a 10-year history of a black macule on her left foot, which had gradually increased in size. Physical examination revealed an irregularly pigmented macule of 45×25 mm on the base of her left hallux (Fig. 1a), which had a parallel ridge pattern and asymmetry of colours on dermoscopy (Fig. 1b). Bilateral erythematous hyperkeratotic lesions were also noted on the palms and soles, showing transgrediens and a whitish spongy appearance soon after water exposure. Her siblings presented with similar hyperkeratotic lesions, whereas her parents were not affected. The findings led to the clinical diagnosis of MM in situ and NPPK.
Fig. 1. (a) A black macule with colour variegation at the base of the left hallux that had gradually increased in size. Diffuse erythema and hyperkeratosis are noted on palmoplantar skin. (b) The macule shows a parallel ridge pattern and asymmetry of colours under dermoscopy. (c) The patient carries the homozygous nonsense mutation c.796C>T (p.Arg266Ter) in SERPINB7. (d) The grafted skin shows no remarkable change after 15 min water exposure, whereas the surrounding affected skin shows a whitish spongy appearance.
The black macular lesion was excised with a 5-mm margin. The wound was reconstructed using a full-thickness skin graft from the right abdomen. Histology revealed the proliferation of atypical melanocytes that were positive for Sox 10, Melan A, and HMB-45 in the epidermis. Parakeratosis limited to the lower layers of the stratum corneum, which is characteristic of NPPK (2), was also noted. To confirm the diagnosis of NPPK, mutation analysis of SERPINB7 was performed. Brief-ly, genomic DNA was extracted from peripheral blood using the QIAamp DNA Blood Maxi Kit (Qiagen, MD, USA) and analysed by Sanger sequencing, as described previously (2). The patient provided written informed consent to participate in this study, in compliance with the Declaration of Helsinki. The Institutional Review Board at the Hokkaido University Graduate School of Medicine approved this study (project No. 14-063). Mutation analysis led to identification of the homozygous nonsense mutation c.796C>T (p.Arg266Ter) (Fig. 1c). These findings verified the diagnosis of MM in situ and NPPK.
The co-existence of NPPK and MM in the present case raises the question of whether NPPK predisposes to MM. To our knowledge, 20 cases of MM complicated by several forms of PPK, such as Papillon-Lefèvre syndrome, Olmsted syndrome, epidermolytic PPK, striate PPK, Mal de Meleda, and NPPK, have been reported (3–8). Of these, it is notable that 8 cases (7 Japanese and 1 Chinese) were complicated by NPPK (4). According to the Human Genetic Variation Database and the 1000 Genomes browser, however, the frequency of mutant alleles carrying the most prevalent founder mutation, c.796C>T, is 0.091 in the Japanese population and 0.0152 in the Chinese population. Therefore, the estimated combined number of NPPK patients in these countries would be more than 0.3 million (9). Given that the estimated prevalence of MM in Japan is 0.0031% based on a national survey in 2014, there appears to be no significant difference in the incidence of MM between the patients with NPPK and the general population. Further studies are needed to address the causal link between NPPK and MM.
It was also noteworthy that the grafted skin showed no NPPK phenotype at 6-month follow-up after surgery (Fig. 1d). Notably, Sartore et al. (3) reported a patient with Mal de Meleda, a disease caused by biallelic loss-of-function mutations in SLURP1, who developed MM on the lesional skin of his hand and underwent complete resection of MM followed by full-thickness skin graft from the inguinal region. Similar to the present case, the grafted skin remained unaffected by PPK even at the 4-year follow-up. Although SERPINB7 and SLURP1 are expressed ubiquitously by differentiated keratinocytes on the whole body (1, 3), these findings suggest that keratinocytes may possess a stable intrinsic regional identity as suggested previously (10) and that the disease-causing effect of mutations in SERPINB7 and SLURP1 may be confined to lesional keratinocytes.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize