


1Department of Dermatology, Peking University First Hospital, 8 Xishiku St, Beijing 100034, and 2Beijing Key Laboratory of Molecular Diagnosis on Dermatoses, Beijing, China. *E-mail: zhimiaolin@bjmu.edu.cn
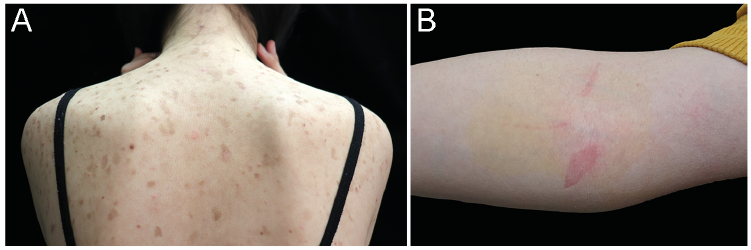
A 13-year-old girl presented to the dermatology clinic with generalized asymptomatic skin scaling since early childhood. The lesions were pink to brown, irregularly shaped macules, 1–2 cm in diameter, disseminated prominently on her trunk (Fig. 1A) and extremities. The lesions were induced or exacerbated by mechanical friction. Erythema occurred after removing tape from the skin (Fig. 1B). Her palms and soles, hair and nails were spared. She was born without obvious abnormalities including bodyweight, skin disorders or other diseases at term after an uneventful pregnancy. No history of atopy was reported, and her family history was unremarkable. A punch biopsy specimen was obtained from her dorsal skin.
What is your diagnosis? See next page for answer.
Fig. 1. Clinical photography. (A) Diffuse pink to brown macules with crusts and scaling on the patient’s trunk. (B) Superficial peels after removing tape from the right cubital fossa.
Acta Derm Venereol 2019; XX: XX–XX.
Diagnosis: Generalized non-inflammatory peeling skin syndrome
Histopathological examination revealed a cleft beneath the stratum corneum with no hyperkeratosis, acanthosis or acantholysis (Fig. 2A). No noticeable infiltration of inflammatory cells was found. A diagnosis of generalized non-inflammatory peeling skin syndrome (PSS) was suspected. Sanger sequencing of the exonic regions of the CDSN, CHST8 and FLG2 genes was performed, which revealed compound heterozygous pathogenic mutations (c.979delC, p.H327Mfs*110; c.1099G>T, p.G367*) in FLG2 (OMIM 618084), confirming the diagnosis of PSS (Fig. 2B).
PSS is comprised of a group of genodermatoses characterized by shedding of the outer epidermis, sometimes accompanied by erythema or blistering. The pathognomonic feature is separation of the epidermis between the stratum corneum and the stratum granulosum without inflammation (1). The 2 major forms of PSS are acral PSS and generalized PSS, the latter of which is subclassified into a non-inflammatory type (type A) and an inflammatory type (type B). Type B is associated with pruritus and atopy (2). Differential diagnoses include epidermolysis bullosa acquisita, epidermolysis bullosa simplex and ichthyosis bullosa of Siemens, which could easily be differentiated according to the clinical and histopathological features.
To date, mutations in 9 genes have been reported to cause PSS. These genes encode proteins that are essential for proper cornification and integrity of the stratum corneum, including components of corneodesmosomes (CDSN, DSG1, FLG2), the cross-linking enzymes (TGM5), and the protease or their inhibitors (LEKTI, CSTA, CAST, or SERPIN8). Among these, 4 genes (CHST8, CAST, SERPINB8, FLG2) are responsible for generalized non-inflammatory PSS. PSS caused by CHST8 mutations shows generalized white scaling, most prominent over the upper and lower limbs (3). PLACK syndrome caused by CAST mutations consist of leukonychia, acral punctate keratoses, cheilitis, and knuckle pads (4). Mutations in SERPINB8 lead to desquamation of the palmoplantar skin, forearms and legs, with some patients demonstrating diffuse palmoplantar keratoderma (5). PSS caused by FLG2 mutations exhibits peeling lesions on the trunk and limbs at sites of minor trauma (6, 7). There is currently no effective treatment for PSS. The patient was advised to avoid trauma and friction.
Fig. 2. (A) Histologic examination of the dorsal skin revealed a separation in the stratum corneum with no obvious inflammatory cells. (Haematoxylin-eosin staining, original magnification ×100). (B) Sanger sequencing revealed compound heterozygous mutations (c.979delC and c.1099G>T) in the FLG2 gene of the patient. Arrows indicate the mutations.
This work was supported by National Natural Science Foundation of China (grant no. 81573032 to Z.L.).


 Click to show fullsize
Click to show fullsize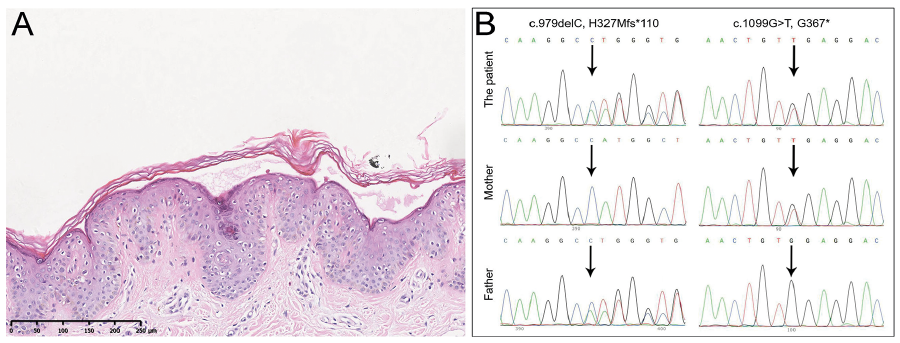 Click to show fullsize
Click to show fullsize