


1Department of Dermatology, University of Montpellier and INSERM U1058, Hôpital Saint-Eloi, 80 avenue Augustin Fliche, FR-34295 Montpellier Cedex 5, 2Department of Dermatology, University of Reims, Reims, and 3Laboratory of Pathology, University of Montpellier, Montpellier, France. *E-mail: o-dereure@chu-montpellier.fr
Accepted Oct 14, 2019; Epub ahead of print Oct 14, 2019
Acta Derm Venereol 2020; 100: adv00035
Primary cutaneous gamma-delta T-cell lymphoma (PCGDTCL) is a rare form of primary cutaneous lymphoma characterized by a clonal proliferation of mature γ/δ T lymphocytes. PCGDTCL is usually considered an aggressive and rapidly evolving malignancy. Contrasting with this patter, we hereby report 2 unusual cases with a protracted indolent evolution.
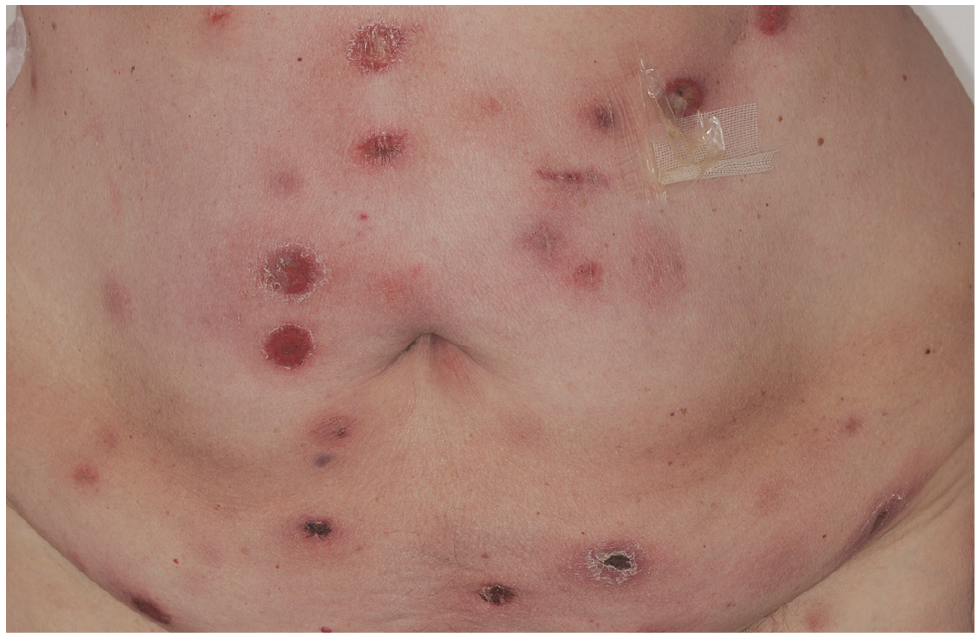
Patient 1. A 68-year-old woman presented with relapsing ulcerated skin nodules, treated with several courses of antibiotics during the preceding 6 years, along with more recent fever and night sweating. Initial physical examination revealed multiple subcutaneous ulcerated nodules located on her limbs and trunk (Fig. 1) some of them evolving toward spontaneous regression according to the patient. A picture of an earlier lesion was obtained retrospectively, showing a thick ulcerated plaque clinically identical to later lesions thus confirming a long initial indolent course. No superficial lymphadenopathy was present and general status was conserved. Histopathological examination of a nodule revealed a dense dermal and subcutaneous infiltrate of medium-to-large lymphocytes displaying a CD3+ CD4– CD8– CD56+ EBER granzyme B+ TIA1+ TCRβ phenotype. Staging with positron emission tomography (PET) showed no suspect hypermetabolism of internal organs and deep lymph nodes. Based on these data, a diagnosis of PCGDTCL was established and a first-line treatment with methotrexate 20 mg/week was introduced owing to the protracted indolent evolution, initially associated with prednisone on account of biological elements of macrophage activation syndrome (MAS) with no related clinical symptoms. However, systemic chemotherapy with cyclophosphamide, adriamycin, vincristin and prednisone (CHOP) was rapidly implemented due to the uncontrolled progression of skin lesions but had to be interrupted after 4 courses owing to infectious complications. The patient finally presented with a deteriorated general condition, dyspnea and relapse of MAS and died one month after the last cycle of chemotherapy.
Patient 2. A 69-year-old man was referred for evaluation of recurrent, self-regressive cutaneous nodules of a 2-year duration, with no significant extracutaneous manifestations. Initial biopsies were consistent with primary cutaneous CD30+ lymphoproliferation, and cutaneous lesions were treated with topical steroids. After 2 years of relapsing and remitting lesions, he finally presented with non-regressive subcutaneous nodules on the legs and trunk. Histopatho-logical examination then revealed a dense predominant dermal and subcutaneous infiltrate of small to medium-size lymphocytes (Fig. S1) expressing a CD3+ CD4– CD8– CD56– granzyme B+ TIA-1+ TCRβ– TCRγ+ phenotype (Fig. S2), whereas GeneScan analysis identified a dominant T-cell population in skin lesions. PET did not display any hypermetabolism suspect of extracutaneous involvement. A diagnosis of PCGDTCL was retained and a treatment with methotrexate 15 mg/week was started. Since no improvement was obtained after 5 months, pegylated liposomal doxorubicin was subsequently introduced replaced after 7 cycles by polychemotherapy with CHOP owing to iatrogenic peripheral neuropathy. This latter therapy was efficient and remained well tolerated after 9 cycles (7 CHOP and 2 COP) with no worsening of peripheral neurotoxicity. No skin progression nor extracutaneous involvement was observed 24 months after the last course of chemotherapy and more than 5 years after the first occurrence of cutaneous nodules.
Fig. 1. Patient 1: multiple ulcerated nodules on the abdomen.
PCGDTCL is a rare and usually aggressive malignancy, accounting for less than 1% of primary cutaneous lym-phomas overall. Patients usually present with sometimes ulcerated or necrotic panniculitis-like plaques, nodules and tumours, mainly located on the limbs and the trunk (1). Histopathological and immunohistochemical analysis typically display an atypical lymphoid infiltrate, involving the epidermis, dermis, and/or subcutis, consisting of CD3+ CD4– CD5– TCRβ– TCRγ+ CD8– medium to large-size cells with variable expression of CD7 and CD56 and a strong expression of the cytotoxic markers granzyme B and TIA-1 (2). This entity usually displays an aggressive clinical course with a median overall survival of 15 months (2), the frequent occurrence of MAS and a poor response to treatment including systemic polychemotherapy (3).
However, an increasing corpus of reports suggests the existence of a less frequently encountered subset with an allegedly more “indolent” or “smouldering” clinical course, variable clinical and histological presentations (4–12) and an overall survival that may range from 2 (4) to more than 15 years (10). Of note, the accurate nature of this “indolent” behaviour appears relatively ambiguous in earlier reports, since it may reflect variable underlying considerations including a less-than-expected clinical aggressiveness, a protracted overall survival (usually estimated since first occurrence of skin lesions) and/or a more favourable response to treatment. More specifically, 18 of such reported cases showed an overall survival exceeding twice the general median figure of 15 months and our 2 cases are consistent with these data (survival of more than 6 and 5 years, respectively, after first occurrence of skin lesions).
In this particular subset, regardless of its likely heterogeneity, clinical presentation appears to be similar to the main, aggressive subset with nodules, indurated plaques, ulcers or blistering lesions (11) located mainly on the limbs and trunk but which may also affect the face (5, 11) or display a widespread distribution. Interestingly Merrill et al. (13) suggested that a predominantly epidermotropic pattern might be associated with a more favourable outcome, although most other reported cases of “indolent” PCGDTCL displayed a more usual predominant dermal and/or subcutaneous involvement as in our 2 patients, even though a sparse epidermotropism was occasionally observed in patient 2. Atypical expression of CD30, CD4 or TCRβF1 might also be associated with a more indolent course (7), a data consistent with CD30 expression and an initial misdiagnosis of primary cutaneous CD30+ lymphoproliferation in our second patient.
This “smouldering” subset PCGDTCL must be separated from lupus erythematosus panniculitis and panniculitis-like subcutaneous T-cell lymphomas, although they may share some clinical and pathological features, including an initially indolent outcome. Additional differential diagnoses include other primary cutaneous T-cell lymphomas and a number of reactive conditions with unusual expression of TCRγ, such as mycosis fungoides (14), primary cutaneous CD30+ lymphoproliferations (15) and pityriasis lichenoides et varioliformis acuta (16).
Long-term outcome in this particular subset is still uncertain, as the overall survival may widely vary across reported cases. Despite the report of protracted complete remissions in several cases (5–7), most authors emphasize that the large majority of patients with so-called smouldering PCGDTCL eventually evolve toward a more usual aggressive form after an initial indolent course with fatal evolution occurring up to 15 years after initial diagnosis (10). These findings are consistent with our first patient’s outcome and clearly raise the issue of considering an (relatively) early, aggressive treatment in some patients during the initial, indolent phase of the disease as in our second case, taking into account age, comorbidities and general status even though there is currently no efficient way to anticipate final outcome on an individual basis. However, such a strategy must be discussed on a case-by-case basis since it is questionable in the most fragile patients owing to possible unnecessary morbidity and mortality induced by toxic treatments that may include hematopoietic stem cell transplantation (17). In all cases, close clinical and morphological monitoring remains mandatory to quickly identify a shift to a more “classical”, aggressive form.


 Click to show fullsize
Click to show fullsize