


1Department of Pathology, Zealand University Hospital, Roskilde, Denmark, 2Department of Dermatology, University Hospital, Schleswig-Holstein, Kiel, Germany, 3Department of Dermatology, Aarhus University Hospital, Aarhus, 4Department of Dermatology, Bispebjerg and Frederiksberg Hospital, Copenhagen, 5Department of Dermatology, Zealand University Hospital, Roskilde, and 6Leo Foundation Skin Immunology Research Center, Department of Immunology and Microbiology, University of Copenhagen, Copenhagen, Denmark
Diagnosis of mycosis fungoides and Sézary syndrome can be very challenging. Clinical and histopathological data for patients with mycosis fungoides and Sézary
syndrome in Denmark are limited. A retrospective study was performed in Region Zealand, Denmark from 1990 to 2016. A total of 43 patients with mycosis fungoides or Sézary syndrome were identified during the period. At the time of diagnosis the patients’ mean age was 64.3 years and 74.5% had early-stage (≤IIA) disease. The mean time from onset of skin disease to diagnosis was 4.4 years. Surprisingly, 43% progressed to a higher disease stage, and risk of disease progression was higher for stage IB than IA (p = 0.01). All cases displayed some degree of epidermotropism and the infiltrates consisted of pleomorphic lymphocytes with a T-helper (CD4+/CD8–) phenotype. This study describes, for the first time, all aspects of clinical and histopathological findings in patients with mycosis fungoides and Sézary syndrome in a well-characterized Danish cohort.
Key words: mycosis fungoides; Sézary syndrome; cutaneous T-cell lymphoma; non-Hodgkin lymphoma.
Accepted Oct 16, 2019; E-published Oct 17, 2019
Acta Derm Venereol 2020; 100: XX–XX.
Corr: Pia Rude Nielsen, Department of Pathology, Zealand University Hospital, Sygehusvej 9, DK-4000 Roskilde, Denmark. E-mail: piarude@dadlnet.dk
Clinical and histopathological data on the characteristics of patients with mycosis fungoides and Sézary syndrome in Denmark are limited. This retrospective study describes the epidemiological, clinical and histopathological features of 43 patients with mycosis fungoides and Sézary syndrome in the eastern part of Denmark during 1990 to 2016. Mean age and clinical stage at the time of diagnosis are in line with similar studies, but, surprisingly, 43% of the patients progressed to a higher disease stage. The risk of disease progression was higher for stage IB than IA.
Cutaneous T-cell lymphoma (CTCL) represents a complex group of disorders with various clinical manifestations, outcomes and therapeutic considerations.
Mycosis fungoides (MF) is the most common type of CTCL. MF represents more than 50% of all primary cutaneous lymphomas, and the reported incidence is up to 6.4 per million people per year in US (1). In early-stage disease, which can last for several years, MF presents as erythematous skin patches and/or plaques, often resembling benign inflammatory conditions. For many patients, the disease never progresses beyond this point; however, up to one-third of patients develop ulcerating tumours or erythroderma, and possible further dissemination of MF to the lymphoid system, blood and internal organs (2, 3).
Sézary syndrome (SS) is a rare subtype of CTCL, which is often considered as the leukaemic variant of MF. SS is defined as a combination of lymphadenopathy, exfoliative/pruritic erythroderma and circulating neoplastic T cells in the blood (2). The clinical and histological presentation of MF/SS can be challenging due to similarities with several benign inflammatory diseases (4). The histopathological findings in MF are heterogeneous and can vary between patients and within the same patient (5, 6). In keeping with this, the diagnostic work-up is often prolonged and several biopsies are needed before a final diagnosis is reached (7). A diagnostic algorithm proposed by Pimpinelli et al. (8), and reviewed by Vandergriff et al. (9) based on clinical, histopathological, immunophenotypical and molecular parameters has been proposed to add diagnostic accuracy in early-stage disease. The aim of the present retrospective study was to characterize the clinical, epidemiological, histological, immunophenotypical and molecular findings, as well as therapeutic regimens, in patients with MF or SS, diagnosed in the region of Zealand in the eastern part of Denmark. In addition, differences in progression free-survival (PFS) between stages IA and IB were analysed.
Patients and clinical data
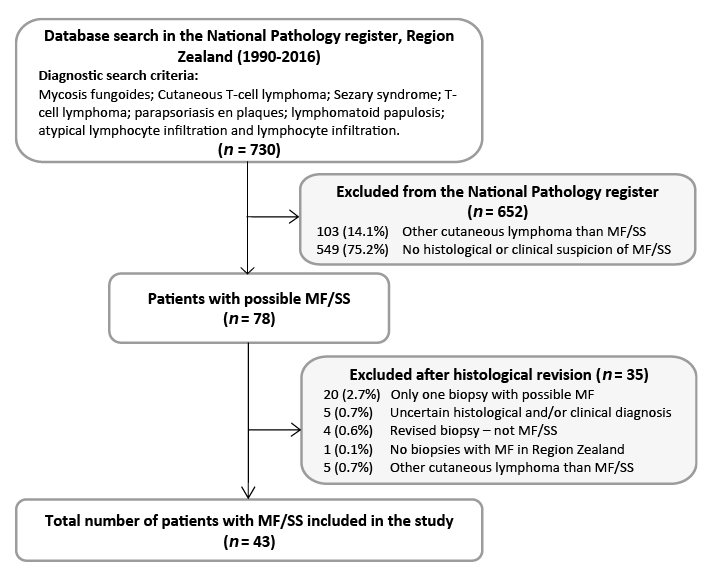
This study is a retrospective, descriptive single-centre study of patients diagnosed with MF and SS in the region of Zealand, Denmark. From the National Pathology Register (Patobank), all patients who had been registered in Region Zealand from January 1990 to December 2016, with a histological diagnosis of MF, SS, CTCL, T-cell lymphoma, parapsoriasis en plaques, lymphomatoid papulosis, atypical lymphocytic infiltration and lymphocyte infiltration were identified. Overall 78 patients with a putative diagnosis of MF or SS were found. After a systemic review of histological slides and clinical records 43 patients with histological and clinically verified MF and SS were identified (Fig. 1). The inhabitants of Region Zealand (currently 835.000) correspond to approximately one-sixth of the Danish population (currently 5.7 million). Clinical information regarding age, sex, clinical stage at the time of diagnosis, disease progression, treatment, co-morbidities, secondary malignancy and follow-up time were registered. The study was approved by the Regional Ethics Committee (SJ-603) and the Danish Data Protection Agency (REG-009-2017).
Fig. 1. Flowchart of inclusion and exclusion of patients in the study cohort.
All patients were staged according to the latest European Organization of Research and Treatment of Cancer (EORTC) and International Society for Cutaneous Lymphomas (ISCL) classification in tumor-node-metastasis-blood and clinical stages (10). The initial skin biopsy was used for histopathological evaluation, but this biopsy did not necessarily represent the diagnostic biopsy in all cases.
Histopathological and immunophenotypical evaluation
Haematoxylin and eosin (H&E)-stained slides from formalin fixed paraffin-embedded (FFPE) skin biopsies were re-evaluated for predefined histopathological features. The histological evaluation was performed blinded with regards to the clinical data. The histological assessment was performed by trained pathologists following international accepted morphological recommendations of MF (5, 8). The following histological criteria were: epidermotropism (scored as none, minor, moderate and strong), Pautrier’s microabscesses (yes/no, defined as presence of ≥ 3 lymphocytes in epidermis), basal alignment of T-lymphocytes visualized by CD3 (yes/no, defined as ≥ 4 lymphocytes next to each other in the basal epidermis), ulceration of epidermis (yes/no), spongiosis (yes/no), folliculotropism (yes/no), infiltrate density (scored as low: a few perivascular and/or dispersed lymphocytes in the superficial dermis; moderate: neither low nor dense; dense: a band-like lymphocytic infiltrate), infiltrate depth (scored as superficial, dermal, subcutaneous), infiltrate pattern (scored as band-like, perivascular or both), presence of atypical lymphocytes (epidermis, dermis or both) and large cells count (defined as twice the size of a normal lymphocyte; scored as < 10%, 10–50%, > 50%).
Due to the retrospective design of the study, it was only possible to access already available immunohistochemical slides. All immunostains performed on the first biopsy were evaluated and scored semiquantitatively by visual inspection. The expression of CD3, CD4, CD5, CD7, CD8, CD20 and CD79a were scored in 4 categories: no/minor expression (≤ 25% positive cells), low expression (26–50% positive cells), moderate expression (51–75% positive cells) and high expression (>75% positive cells). CD30 was slightly differently scored in 4 categories: no/minor expression (≤ 5%), low expression (6–20%), moderate expression (21–50%) and high expression (> 50%). The score was performed as percentage of positive lymphocytes in the population of all lymphocytes. The Ki-67 proliferation index was counted in hotspots of 200 positive lymphocytes (magnification ×400) in the dermal compartment, excluding counting the lymphocytes in hair follicles, sweat glands and vessels.
T-cell receptor (TCR) gene re-arrangement analysis was performed according to the Biomed-2 protocol (11).
Statistical analysis
Statistical analysis was performed using SPSS version 24 software (IBM Corporation, Armonk, NY, USA). Baseline characteristics were described using median, standard deviation (SD) and means with ranges for the continuous variables, and numbers with percentage frequencies for the categorical variables. PFS curves were calculated according to the Kaplan–Meier method and compared using the log-rank test. Analyses of PFS, first disease progression (>IIA) or MF-related death were considered as an event.
Patient demographic and clinical characteristics
Forty-three patients with clinically and histologically verified MF or SS were identified during the period 1990 to 2016 in Region Zealand, Denmark. All data regarding demographics, clinical presentation and follow-up are summarized in Table I. The mean age at the time of diagnosis was 64.3 years (range 31–83 years) and the male to female ratio was almost equal (1:1.1). At the time of diagnosis 32 (74.4%) of the patients were diagnosed in early-stage disease (IA–IIA), and advanced-stage disease (IIB–IVB) was encountered in only 7 patients (16.3%) at the time of diagnosis. In 4 patients, the disease stage could not be specified due to lack of data in the medical record. Clinical and histological variants of MF included classical MF (86.1%), folliculotropic MF (2.3%) and erythrodermic MF (2.3%). Four patients (9.3%) were diagnosed with SS. Time from the reported onset of skin disease to the diagnosis ranged from 0.25 to 23 years (mean 4.4 years). The mean number of biopsies to establish the diagnosis was 3.14, ranging from 1 to 9 biopsies. Mean follow-up time from diagnosis to last follow-up was 7.5 years (range 0.5–33 years). Almost half (43%) of the patients experienced disease progression and the mean time to progression was 47.7 months (range 1–168 months). Thirteen patients (30.2%) were dead and 7 (16.2%) of these died of MF-related infections. Other causes of death were primarily due to other malignancies. Our study population exhibited a wide range of co-morbidities, with cardiovascular and endocrinological diseases being the most prevalent. Three patients experienced hypothyroidism as a side-effect to the lymphoma treatment. None of the patients had HIV or chronic inflammatory diseases. Overall, 16 patients (37.2%) had or were later diagnosed with a secondary malignancy. Patients primarily had basal cell carcinoma (BCC) and squamous cell carcinoma (SCC) of the skin. Two patients were diagnosed with SCC, one patient with BCC and one patient with both BCC and SCC in the phototherapy-treated areas. Two patients were diagnosed with either BCC or SCC prior to the diagnosis of MF. Other cancers included soft-tissue sarcoma, breast, lung, bladder and uterine carcinoma and other haematological cancers (Table I). There was no report of melanoma in the cohort.
Table I. Demographic and clinical characteristics of 43 patients with mycosis fungoides and Sézary syndrome
Initial and overall treatment in the study cohort
An overview of the initial treatment modalities is shown in Table I. Topical steroids were given at the time of the primary biopsy in 16 (32%) cases. Phototherapy, ultraviolet B (UVB) and psoralen plus ultraviolet A (PUVA) alone, or in combination with topical corticosteroids, were used in 4 patients with clinical stage IA or IB. An erythrodermic patient (stage III) received a combination of UVB and topical steroids as the initial treatment, and she subsequently received methotrexate with full remission. One patient was initially misdiagnosed with granulomatosis with polyangiitis and therefore treated with cyclophosphamide. In summary, the different therapy regimens included topical treatments with corticosteroids and pimecrolimus, UVB, PUVA, prednisolone, localized radiation therapy, total skin electron beam (TSEB) radiation, photopheresis, interferon-α, bexarotene, mycophenolic acid, azathioprine, cyclosporine, acitretin, carmustine, systemic chemotherapies (methotrexate, R-CHOP and CHOEP). Finally, one patient was treated with allogenic haematopoietic stem cell transplantation. Unfortunately, the patient died of graft versus host disease.
Histopathological findings
The histopathological findings of the first skin biopsies are shown in Table II. In 14/43 (32.6%) of cases the first biopsy was diagnostic for MF.
All biopsies displayed some degree of epidermotropism and, to a minor degree, in most cases 28/43 (65.1%). Pautrier’s microabscesses were present in 16/43 (37.2%) and basal alignment were seen in 17/43 (39.5%) of the cases. In all primary biopsies, the infiltrate constituted of small-medium pleomorphic lymphocytes with few or no larger cells (< 10%). In sequential biopsies, 2 patients developed large cell transformation. Twenty-eight cases (65.1%) revealed a superficial dermal infiltrate and the remaining 15 (34.9%) displayed a deeper dermal infiltrate. Mean infiltrate thickness was 0.74 mm (range 0.12–2.8 mm). The infiltrate pattern of infiltration was band-like in 10 cases, perivascular in 8 cases, and a combination of both in 25 biopsies. The majority of cases (83.7%) presented with low or moderate infiltrate density. Seven cases (16.3%) displayed a dense infiltrate pattern in their primary biopsy. Follicular epitheliotropism/exocytosis was seen in 11 out of 16 cases. In 27 cases, folliculotropism could not be evaluated due to lack of hair follicles. The original histological diagnoses of the first skin biopsy in Region Zealand were distributed as MF (27.9%), suspicious for MF (16.3%), non-specified dermatitis (37.2%), atypical lymphocytic infiltration (9.3%), parapsoriasis en plaques (2.3%), lymphomatoid papulosis (2.3%) and guttate psoriasis (4.7%).
Table II. Histopathological characteristics in the first available biopsy of 43 patients with mycosis fungoides and Sézary syndrome in Region Zealand
Immunohistochemical findings
Immunohistochemical staining revealed in 15/15 (100%) tested cases a T-helper phenotype with CD4+/CD8–. The pan T-cell marker CD3 was preserved in 40/40 (100%) tested cases; however, a partial loss of CD5+ and CD7+ was observed in 1/15 (6.7%) and 6/8 (75%) tested cases, respectively. B cells were present in 20/20 (100%) tested cases (range 10–30% of the infiltrate). One case showed a high degree of (>75%) of CD20+ cells. These B cells were both diffusely distributed and/or seen in small nodular infiltrates in dermis. Scattered CD30+ cells were observed in 13/14 (92.9%) tested cases, all at a percentage lower than 50%. Proliferation rate, indicated by Ki-67, was ≤ 50% in the tested cases (n = 7).
T-cell receptor clonality findings
This study has been conducted retrospectively and data regarding TCR gene rearrangement were available in only 21/43 (48.8%) of the patients. TCR clonality was detected in 20 patients, with one patient having an inconclusive result.
Progression-free survival between stage IA and IB
PFS was compared between patients in stages IA and IB. PFS was defined as time to first disease progression (>IIA) or death due to MF. The median time to disease progression was 8.0 years for all patients in stage IA or IB. The risk of disease progression was significantly higher for stage IB compared with stage IA (p = 0.01) (Fig. 2). In the overall follow-up time 27.8% (5/18) of patients with stage IA and 71.4% (10/14) with stage IB progressed to a higher disease stage than IIA.
Fig. 2. Kaplan–Meier curve for progression-free survival (PFS) for stages IA (n = 18) and IB (n = 14) in the study cohort. *Log-rank test.
This is the first Danish study to describe patients with MF and SS from initial biopsy to final follow-up in a single centre. The mean patient age in this study was 64.3 years, which is a little higher than reported previously (12, 13). In addition, the current study found a slightly higher prevalence of women diagnosed with MF, compared with most epidemiological reports claiming a male predominance (12, 14, 15). In agreement with a similar study from Sweden, mean time from onset of skin disease to established MF diagnosis was 4.4 years. Likewise, the distribution of clinical stage at initial diagnosis of MF was quite similar (16). Surprisingly, 43% of our study cohort progressed to a higher disease stage. This is in conflict with similar studies where disease progression is reported in approximately one-third of patients (13, 16, 17). A large study from the USA found that only 11.6% of their patients progressed to a higher disease stage (12). In Denmark, all biopsies in the diagnostic work-up and monitoring of patients are recorded in the National Pathology registry (Patobank). In addition, medical records are available for physicians involved in patient management and clinical progression, e.g. tumour development is recorded in the medical records, but unfortunately not always biopsied. In keeping with this, a false tumour-stage MF may be recorded in the medical journal. Also, any variation in numbers has a profound impact in our small study cohort. In 2 patients, disease progression date was absent, which might result in a slightly increased progression percentage in our cohort. There is currently no uniform definition for disease progression in CTCL (18, 19).
As expected, time to disease progression in the current study was lower for patients in stage IB than IA. This reflects previous observations, where risk of disease progression is associated with the severity of the initial disease stage (14, 15). Clinical prognostic factors, such as patient’s age, sex, extracutaneous disease, T-classification, large cell transformation and elevated lactate dehydrogenase (LDH) at the time of diagnosis are considered and validated as prognostic factors (12, 14, 15). Unfortunately, LDH serum levels were not taken in the diagnostic work-up in the majority of our patients and therefore no conclusion could be drawn regarding LDH at the time of diagnosis. None of the patients had large-cell transformation at the time of diagnosis. A recently published study by Lindahl et al. (20) identified a miRNA classifier as an effective prognostic tool in detecting patients with early-stage MF at risk of progression. Moreover, Wehkamp et al. (21) demonstrated that clinically defined subgroups of MF display different histological features at the initial biopsy. Patients with later progression to a higher disease stage appear to have denser and deeper infiltrates with a higher proliferative index and a higher amount of large cells. However, the impact for the individual prognosis, based only on the histopathological analysis, seems to be limited.
The most common histopathological diagnosis preceding MF in our study cohort was a non-specific dermatitis and atypical lymphocytic infiltration. One patient was initially diagnosed with parapsoriasis en plaques and another with lymphomatoid papulosis. Previous studies have shown that parapsoriasis en plaques have the potential to develop into MF (22) and lymphomatoid papulosis is associated with a higher risk of developing MF (23). The diagnosis of early MF can be very challenging for both clinicians and pathologists, and an accurate diagnosis depends on specific knowledge and long-term experience on both parties.
The histopathological diagnostic criteria of early MF was defined by Kerl & Kresbach (24) and Sanchez & Ackerman (25), and have been validated and refined by several subsequent larger studies (26–28). Epidermotropism is considered the histopathological hallmark of MF and has been reported in >75% of cases of MF (5, 26, 27). In our study, some degree of epidermotropism was observed in all cases and Pautrier’s microabscesses were seen in 37.2%, together with basal alignment of lymphocytes in 39.5%. These findings are in agreement with previous studies (26, 28). In the majority of cases, the dermal infiltrate consisted of a superficial lymphohistiocytic infiltrate composed of small-to-medium-sized lymphocytes with a T-helper phenotype (CD4+/CD8–). The infiltrate density was mild to moderate and in more than half of the cases the pattern were both band-like and perivascular. This infiltrate pattern in the first biopsy is consistent with previous findings in initial biopsies of patients with MF (21). In almost 60% of the biopsies, some degree of spongiosis was present. In the cases with spongiosis in combination with a minor degree of epidermotropism and lack of Pautrier’s microabscesses, the primary pathologist settled on a diagnosis of unspecified dermatitis, which is a well-known phenomenon in diagnosing early-stage MF (5, 29). The majority of patients presenting with dermatitis-like skin symptoms are often treated with topical steroids and the biopsy is taken upon treatment failure. Topical steroids dampen the inflammation, thereby to some extent obscuring the histological findings in early-stage MF. First-line treatment in our cohort consisted in 42% of the patients of topical steroids alone or in combination with PUVA or UVB. This is in line with the EORTC consensus guidelines for treatment of MF/SS in early-stage disease (30). Although PUVA was only used as first-line therapy in one patient, just over half (56%) of the patients eventually received PUVA. The risk of developing PUVA-associated skin cancer, especially non-melanoma skin cancer (NMSC), has primarily been investigated in patients with psoriasis (31). The results are pointing toward an increased risk of NMSC (mainly SCC) in patients who are exposed to high doses of PUVA (31). In addition, an increased risk of melanoma in PUVA-treated patients with psoriasis was reported in studies from the USA, but not in Europe (31). In our cohort 6 patients had NMSC as a secondary malignancy and none developed melanoma during the follow-up period. BCC and SCC were the most frequent secondary malignancies and 4 patients eventually developed NMSC in the phototherapy-treated regions. Although 37.2% of our study cohort were diagnosed with a secondary malignancy, this was as expected in this ageing patient cohort and did not exceed cancer rates for this period in the background population (32).
Although this is the first Danish study to fully address clinical, epidemiological and histological data in patients with MF and SS, there are certain limitations that should be emphasized. This study consists of retrospective data, based on a small study cohort from a limited geographical area, and cannot be generalized to other populations. In summary, these results support previous findings and challenges that both clinicians and pathologists experience when working with patients with CTCL. Presentation of MF and SS are both heterogeneous and may mimic a variety of inflammatory skin diseases, resulting in delayed diagnosis, postponed therapeutic interventions, and a need for future research into diagnostic and prognostic biomarkers.
team at the Department of Pathology (Næstved, Denmark) is highly appreciated. The authors would also like to thank Peter Lerche for critical proofreading prior to submission.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize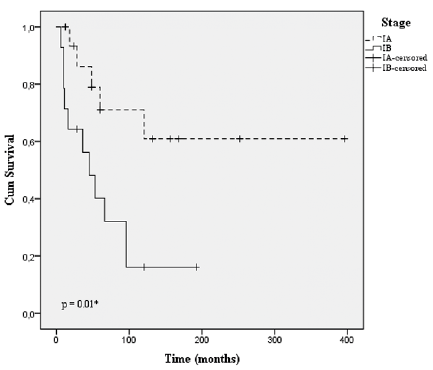 Click to show fullsize
Click to show fullsize