


1Guenther Dermatology Research Centre, London, Ontario, Canada, 2Eli Lilly and Company, Indianapolis, Indiana, USA, 3Medical Dermatology Specialists, Atlanta, Georgia, USA, 4Centre de Recherche Dermatologique du Quebec metropolitain, Quebec City, Quebec, Canada, 5Veracity Clinical Research, Brisbane, Australia, 6Probity Medical Research, Waterloo, Ontario, Canada, 7College of Pharmacy, University of Cincinnati, Cincinnati, Ohio, USA, and 8Charles Institute of Dermatology, University College, Dublin, Ireland
Ixekizumab was efficacious in treating moderate-to-severe genital psoriasis over 12 weeks. We evaluated the long-term efficacy and safety of ixekizumab for up to 52 weeks. Patients were randomized to 80 mg ixekizumab every 2 weeks or to placebo through Week 12, then received 80 mg open-label ixekizumab every 4 weeks through Week 52. In patients initially randomized to ixekizumab, clear or almost clear genital skin was achieved for 73% of patients at Week 12 and 75% at Week 52. Persistent improvements were also observed for overall psoriasis, genital itch, and the impact of genital psoriasis on the frequency of sexual activity. The safety profile was consistent with studies of ixekizumab in patients with moderate-to-severe plaque psoriasis. Ixekizumab provided rapid and persistent improvements in the signs and symptoms of genital psoriasis for up to 52 weeks of treatment.
Key words: ixekizumab; genital psoriasis; IXORA-Q; clinical trial; sexual impact; itch.
Accepted Oct 16, 2019; E-published Oct 17, 2019
Acta Derm Venereol
Corr: Lyn Guenther, Guenther Dermatology Research Centre, 835 Richmond Street, London, Ontario, N6A 3H7, Canada. E-mail: dgue@guenthermpc.com
Genital skin is commonly affected among patients with psoriasis. Genital psoriasis can cause problems such as genital itch and pain, and have negative effects on mental, social, and sexual health. Ixekizumab, can provide significant relief of genital psoriasis within 12 weeks of starting treatment. This study showed that patients with genital psoriasis continue to have symptom relief after one year of treatment with ixekizumab.
Psoriasis is a chronic, immune-mediated skin disease with a prevalence of approximately 3% in adults in the United States (1). Among patients with psoriasis, up to 63% of patients have genital involvement at some point during the course of their disease (2, 3). Genital psoriasis often presents as well-defined, thin, erythematous plaques without scaling, which can affect both the genital skin and mucosa, and can be accompanied by fissures, ulcers, and/or erosions (4). The impact of genital psoriasis on patients is multidimensional, characterized by bothersome physical symptoms such as genital itch and pain as well as by negative effects on psychological, social, and sexual well-being (5). Genital psoriasis may present to multiple medical disciplines including primary care, dermatology, venereology, gynecology, urology, and sexual medicine (6). Despite the high prevalence and burden of genital psoriasis, approximately 46% of patients do not discuss their genital symptoms with their healthcare providers and only 40% of patients indicate having been examined for genital involvement (7–9). Thus, there is currently an unmet need for increased awareness of genital psoriasis among both patients and physicians to encourage appropriate diagnosis and treatment.
Ixekizumab (IXE), a high-affinity monoclonal antibody targeting IL-17A, is currently the only treatment approved for patients with moderate-to-severe plaque psoriasis that includes information in the labeling (e.g. in the United States, European Union, and Canada) about successful treatment of patients with genital involvement. In IXORA-Q, a Phase 3, randomized, placebo (PBO)-controlled, double-blind clinical trial in patients with moderate-to-severe genital psoriasis, 73% of patients receiving 80 mg IXE every 2 weeks (Q2W) achieved clear or almost clear genital skin compared to 8% of patients receiving PBO (p < 0.001) following 12 weeks of treatment (10). Here, we report the efficacy and safety of IXE through 52 weeks of treatment in patients with moderate-to-severe genital psoriasis.
Participants
Complete eligibility criteria have been previously published (10). Briefly, eligible study participants were adults with moderate-to-severe genital psoriasis who were candidates for phototherapy and/or systemic therapy. At screening and baseline visits, participants were required to have an overall static Physician’s Global Assessment (sPGA) score ≥ 3, an sPGA of Genitalia (sPGA-G) score ≥ 3, body surface area (BSA) involvement of ≥ 1% (up to 40% of enrolled patients could have BSA of 1% to < 10%), and confirmation of plaque psoriasis in a nongenital area. Eligible participants had failed to respond to, or were intolerant of, at least one topical therapy used for treatment of genital psoriasis (corticosteroids, calcineurin inhibitors, or vitamin D analogs) and were required to use a reliable method of birth control during the study. Key exclusion criteria included pustules or vesicles in the genital area, or previous treatment with an IL-17 antagonist. Participants were excluded if they had a medical condition that could pose an unacceptable risk to the patient or that could interfere with interpretation of study results.
Study design
Eligible patients were randomized (1:1) to receive subcutaneous injections of 80 mg IXE Q2W, following a 160-mg starting dose (two 80-mg doses) at Week 0, or to PBO Q2W (following 2 subcutaneous injections of PBO at Week 0) during a 12-week blinded treatment period. At Week 12, patients entered an open-label treatment period, during which all patients received 80 mg IXE every 4 weeks (Q4W) up to Week 52. Patients initially randomized to PBO received a starting dose of 160 mg IXE at Week 12 and patients initially randomized to IXE received one 80-mg dose of IXE plus one dose of PBO. During study visits at Week 24, 28, and 40, patients had an option to increase dosing frequency (which was maintained until Week 52) to 80 mg IXE Q2W, if determined by the patient and investigator to be necessary to achieve or maintain satisfactory disease control.
IXORA-Q was conducted according to the ethical principles of the Declaration of Helsinki. The study protocol was approved by an ethical review board at each participating site. All patients provided written informed consent prior to undergoing study procedures. IXORA-Q is registered on ClinicalTrials.gov (NCT02718898).
Outcomes
The primary and major secondary efficacy endpoints (assessed at Week 12) have been previously published (10). The pre-defined efficacy outcomes assessed through Week 52 included the proportion of patients achieving an sPGA-G score of 0 or 1 (sPGA-G 0/1) or 0 (sPGA-G 0) (11); an overall sPGA score of 0 or 1 (overall sPGA 0/1) or 0 (overall sPGA 0); an improvement of at least 3 points from baseline in the Genital Psoriasis Symptoms Scale (GPSS) (12) itch item (genital itch numeric rating scale [NRS]) among patients with a score ≥ 3 at baseline; the proportion of patients achieving a genital psoriasis Sexual Frequency Questionnaire Item 2 Score of 0 or 1 (GenPs-SFQ Item 2 [0,1]) – indicating that the frequency of sexual activity was never or rarely limited by genital psoriasis – among patients with a baseline score ≥ 2 (13); an improvement ≥ 2 points from baseline in the Patient’s Global Assessment of Genital Psoriasis (PatGA-Genital) among patients who had a baseline score ≥ 2; the mean change from baseline in the modified Genital Psoriasis Area and Severity Index (mGPASI) (3, 14); and the mean change from baseline in the total and each individual item score (itch, pain, discomfort, stinging, burning, redness, scaling, and cracking) of the GPSS. An additional post hoc assessment was performed for the proportion of patients achieving an improvement of ≥ 4 points from baseline in the genital itch NRS among patients with a score ≥ 4 at baseline.
With the exception of the PatGA-Genital, efficacy outcomes used in the IXORA-Q study have been described in detail previously (10). The PatGA-Genital is a patient-reported, single-item measure that asks patients to rate the severity of their genital psoriasis on a numeric rating scale that ranges from 0 (clear, no genital psoriasis) to 5 (severe genital psoriasis). The sPGA-G, overall sPGA, and mGPASI were measured at each study visit through Week 52. The PatGA-Genital was assessed at baseline and at each post-baseline visit through Week 52. The GPSS and GenPs-SFQ were measured using an electronic diary either daily (itch NRS) or weekly (GenPs-SFQ) from screening to Week 12, then at each study visit thereafter through Week 52. For the sPGA-G, GPSS, GenPs-SFQ Item 2, and mGPASI, the genital region was defined as the labia majora, labia minora, and perineum in females or the penis, scrotum, and perineum in males.
The presence (yes/no) of psoriasis on the scalp, nails, face, axillae, inframammary fold, inguinal creases, gluteal cleft, perianal region, and pubis was assessed at baseline and at Weeks 4, 12, 24, and 52. The proportion of patients who achieved complete resolution (absence) of psoriasis affecting each region was determined for patients with presence at baseline.
Photographs of the genital region were collected from consenting patients according to a standardized protocol by trained clinical personnel at select participating study sites. Photographs were taken during baseline and post-baseline visits (Weeks 2, 12, 24, and 52) of patients at the participating sites who elected to participate. For patients who volunteered, an additional informed consent was obtained prior to photographs being taken.
Anti-drug antibodies were assessed using validated assays to determine the presence, titer and neutralizing activity of anti-drug antibodies.
Safety outcomes were evaluated by monitoring of adverse events (AEs), vital signs, laboratory tests, and findings from physical exams. AEs of special interest included cytopenias, liver function test changes, infections, injection-site reactions, allergic reactions/hypersensitivities, cerebrocardiovascular events, malignancies, inflammatory bowel disease, and depression. Data on AEs related to cerebrocardiovascular events or inflammatory bowel disease were adjudicated by external, blinded clinical events committees. Depressive symptoms were measured using the Quick Inventory of Depressive Symptomatology-Self Report (16-items) (QIDS-SR16), and suicidal ideation and behavior was monitored using the Columbia-Suicide Severity Rating Scale (C-SSRS).
Statistical analysis
Efficacy analyses were conducted on all randomized patients according to their initially assigned treatment (intention-to-treat population) through Week 12 for patients randomized to PBO or through Week 52 for patients randomized to IXE at baseline. For PBO-randomized patients, analyses of efficacy following Week 12 were conducted on patients who received at least one dose of IXE during the open-label treatment period (open-label treatment period population).
During the blinded treatment period, treatment group comparisons were analyzed using a logistic regression model with treatment and baseline BSA category (1% to < 10% and ≥ 10%) as factors (10). Long-term efficacy was summarized using descriptive statistics. Missing data were imputed using nonresponder imputation (NRI) for categorical variables or modified baseline observation carried forward for continuous variables. At Week 52, the proportion of patients achieving an sPGA-G score of 0/1 was assessed for patient subgroups stratified by baseline BSA involvement (1% to < 10% and ≥ 10%). Patients with dose adjustment to IXE Q2W during the open-label treatment period were included in these analyses.
The categorical efficacy analysis was also summarized through Week 52 using a modified nonresponder imputation, consisting of combined nonresponder imputation and multiple imputation [MI], also referred to as modified MI. For modified NRI, subjects who discontinued because of AEs or lack of efficacy were considered nonresponders and were imputed as NRI. In all other cases of missing data for this analysis, the data were imputed using the MI method, where missing data were imputed to estimate what observations would have been if the patient had not discontinued participation in the study. Specifically, MI is the partial imputation of nonmonotone missing data using Markov chain Monte Carlo method with the simple imputation model, followed by a sequential regression imputation with the baseline score. This analysis was conducted using 2 patient populations: (1) patients randomized to IXE at Week 0 regardless of dose adjustment, and (2) patients randomized to IXE at Week 0 with dose adjustment where only those visits during which patients were treated with the recommended IXE dosing regimen were included and data collected at visits with IXE dose adjustment to IXE Q2W were excluded before application of any imputations.
Safety outcomes are summarized for all patients who received at least one dose of IXE, herein after referred to as the all IXE exposures population. Baseline was defined as the last available value before the first injection of IXE. Adverse events of allergic/hypersensitivity reactions and injection-site reactions were also summarized for treatment-emergent anti-drug antibody (TE-ADA)-positive patients.
The frequency of TE-ADAs and neutralizing anti-drug antibodies (nADAs) are summarized for anti-drug antibody (ADA)-evaluable patients, defined as patients with an evaluable baseline sample and at least one evaluable post-baseline sample or patients with no evaluable baseline sample whose evaluable post-baseline samples were all ADA-negative. TE-ADA-positive patients were defined as patients with at least a 4-fold increase over a positive baseline ADA titer or, for patients with no ADAs detected at baseline, an increase from the baseline to a titer of at least 1:10. TE-ADA-positive patients were grouped by TE-ADA titer according to their last titer value within the baseline/post-baseline period. Titer groups were defined as low titer (< 1:160), moderate titer (≥ 1:160 and < 1:1280), and high titer (≥ 1:1280). Patients positive for TE-ADAs were defined as nADA-positive if they were positive for nADAs for at least 1 TE-ADA-positive sample.
Of 183 patients screened, 149 were randomized to either PBO (n = 74) or IXE Q2W (n = 75) (Fig. S1). Of all randomized patients, 139 (93%) completed the blinded treatment period and entered the open-label treatment period, and 126 (85%) completed Week 52 assessments. Overall the frequency of discontinuation through Week 52 were similar between treatment arms. Among patients initially randomized to PBO, 12 (16%) discontinued (9 in the blinded treatment period and 3 in the open-label treatment period). Among patients initially randomized to IXE, 11 (15%) discontinued (1 in the blinded treatment period and 10 in the open-label treatment period). Baseline demographics and disease characteristics of IXORA-Q have been previously published and were consistent between treatment arms (Table I) (10, 15).
Table I. Baseline demographics and disease characteristics in IXORA-Q
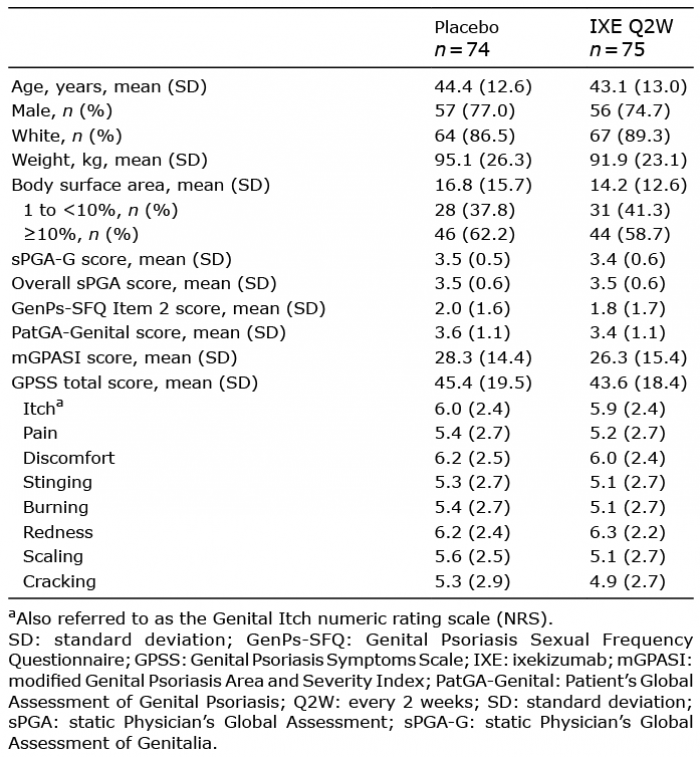
High rates of clinical improvement in genital psoriasis were rapidly achieved and persisted through 52 weeks of treatment. In the patients initially randomized to IXE, clear or almost clear genital skin (sPGA-G 0/1) was achieved by 73% and 75% of patients at Weeks 12 and 52, respectively (Fig. 1a). A similar pattern was observed for complete clearance of genital psoriasis, with sPGA-G 0 response achieved by 56% and 60% of patients at Weeks 12 and 52 (Fig. 1b). Patients initially randomized to PBO also showed rapid improvements in genital psoriasis following treatment with IXE Q4W and achieved sPGA-G 0/1 and sPGA-G 0 responses of 79% and 59% at Week 52, respectively. Response rates for sPGAG 0/1 at Week 52 were consistent between lower (1% to < 10%) and higher (≥10%) BSA subgroups for both treatment arms (Fig. 1c). Rapid and persistent improvement in genital psoriasis was also observed as measured by the mGPASI. Mean (SD) changes from baseline in mGPASI were −22.2 (15.8) at Week 12 and −22.2 (16.0) at Week 52 in patients initially randomized to IXE and −23.4 (14.7) at Week 52 in the PBO/IXE Q4W population.
Fig. 1. Genital psoriasis severity during 52 weeks (NRI) of treatment in IXORA-Q. (a) Proportion of patients achieving a static Physician’s Global Assessment of Genitalia (sPGA-G) score of 0 or 1 at each post-baseline visit. (b) Proportion of patients achieving complete resolution of genital psoriasis (sPGA-G 0) at each post-baseline visit. (c) Proportion of patients achieving a sPGA-G 0/1 at Week 52 among patients with lower (1 to <10%) and higher (≥10%) body surface area involvement at baseline. (d) Proportion of patients achieving at least a 2-point improvement in the Patient’s Global Assessment of Genital Psoriasis among patients with a baseline score of ≥2. (a–d) Results are summarized for the placebo (PBO) population (all patients randomized to placebo at Week 0) through Week 12, the PBO/IXE Q4W population (all patients randomized to placebo at Week 0 who entered the open-label treatment period and received at least 1 dose of ixekizumab) from Weeks 12 to 52, and the IXE population (all patients randomized to ixekizumab at Week 0) through Week 52.
At Week 12, 73% of patients initially randomized to IXE achieved overall sPGA 0/1 and 40% achieved an overall sPGA 0 (Fig. 2). Response rates observed at Week 12 were sustained through Week 52, with 72% achieving overall sPGA 0/1 and 47% achieving overall sPGA 0. Similar response rates were observed at Week 52 for the PBO/IXE Q4W population (overall sPGA 0/1: 74%; overall sPGA 0: 45%).
Fig. 2. Overall psoriasis during 52 weeks of treatment (nonresponder imputation) in IXORA-Q. (a) Proportion of patients achieving an overall static Physician’s Global Assessment (sPGA) score of 0 or 1 at each post-baseline visit. (b) Proportion of patients achieving complete resolution of overall psoriasis (sPGA 0) at each post-baseline visit. (a and b) Results are summarized for the PBO population (all patients randomized to placebo at Week 0) through Week 12, the PBO/IXE Q4W population (all patients randomized to placebo at Week 0 who entered the open-label treatment period and received at least one dose of ixekizumab) from Weeks 12 to 52, and IXE population (all patients randomized to ixekizumab at Week 0) through Week 52.
Rapid and persistent improvement in genital psoriasis was reported for the PatGA-Genital, a patient-reported assessment of genital psoriasis severity (Fig. 1d). Among patients initially randomized to IXE with a baseline PatGA-Genital score ≥ 2, 72% achieved at least a 2-point improvement from baseline at Week 12. At Week 52, PatGA-Genital ≥ 2 point response rates were 74% for patients initially randomized to IXE and 80% for the PBO/IXE Q4W population. Photographs of a female patient and a male patient in Fig. 3 illustrate improvements in genital skin through Week 52.
Fig. 3. Photographic examples of improvement in genital psoriasis during 52 weeks of treatment in IXORA-Q. (a) Photographs of the genital region of a female patient in the PBO/IXE Q4W population. (b) Photographs of the genital region of a male patient in the IXE population.
A total of 52 (35%) patients increased dosing frequency from IXE Q4W to IXE Q2W during the open-label treatment period, including 25 (33%) patients initially randomized to IXE. Among these 25 patients, most had achieved sPGA-G 0–2 (68%), and/or overall sPGA 0–2 (72%), and/or PatGA-Genital 0–2 (60%) at the visit when the decision was made to increase dosing frequency (Table SI). Among patients who received the approved dosing regimen (excluding data from patient visits in which the dose had been increased to IXE Q2W), clinical response rates at Week 52 were 81% for sPGA-G 0/1, 64% for sPGA-G 0, 83% for overall sPGA 0/1, and 54% for overall sPGA 0 (Table SII).
Consistent with improvements in genital psoriasis severity, rapid and persistent improvements through Week 52 were observed in the symptoms associated with genital psoriasis as measured by the mean change from baseline in the total and individual item scores of the GPSS (Table II). Responses in the GPSS at Week 52 were generally consistent between treatment arms. Among patients initially randomized to IXE with a genital itch NRS score of ≥ 3 at baseline, the percentage of patients who achieved at least a 3-point improvement was 60% at Week 12 and 73% at Week 52 (Fig. 4a). Among patients initially randomized to IXE with a baseline genital itch NRS score ≥ 4, 59% and 73% achieved at least a 4-point improvement from baseline at Weeks 12 and 52, respectively (Fig. 4b). Response rates at Week 52 in the PBO/IXE Q4W population for the genital itch NRS (genital itch NRS ≥ 3: 69%; genital itch NRS ≥ 4: 75%) were consistent with those observed in patients initially randomized to IXE.
Table II. Change from baseline in Genital Psoriasis Symptoms Scale (GPSS) through Week 52 of IXORA-Q
Fig. 4. Patient-reported outcomes during 52 weeks (nonresponder imputation) of treatment in IXORA-Q. (a) Proportion of patients achieving at least a 3-point improvement in the genital itch numeric rating scale (NRS) at each post-baseline visit among patients with a score of ≥3 at baseline. (b) Proportion of patients achieving at least a 4-point improvement in the genital itch NRS at each post-baseline visit among patients with a score of ≥4 at baseline. (c) Proportion of patients achieving a score of 0 or 1 in the genital psoriasis Sexual Frequency Questionnaire Item 2 (GenPs-SFQ Item 2), indicating genital psoriasis never or rarely limited the frequency of sexual activity, among patients with a score of ≥2 at baseline. (a–c) Results are summarized for the PBO population (all patients randomized to placebo at Week 0) through Week 12, the PBO/IXE Q4W population (all patients randomized to placebo at Week 0 who entered the open-label treatment period and received at least 1 dose of ixekizumab) from Weeks 12 to 52, and IXE population (all patients randomized to ixekizumab at Week 0) through Week 52.
Among patients with sexual impact at baseline (defined as a GenPs-SFQ Item 2 score of ≥ 2; including 41% of patients in the PBO/IXE Q4W population and 38% of patients initially randomized to IXE), IXE resulted in rapid and persistent improvements in the impact of genital psoriasis on the frequency of sexual activity (Fig. 4c). In patients initially randomized to IXE, 78% and 76% achieved GenPs-SFQ Item 2 0/1 at Week 12 and Week 52, respectively, indicating genital psoriasis never or rarely impacted the frequency of sexual activity. Similar response rates were observed in the PBO/IXE Q4W treatment arm, with 77% achieving GenPs-SFQ Item 2 0/1 at Week 52.
The impact of IXE on complete resolution (absence) of psoriasis was evaluated for multiple body regions among patients with presence in each respective location at baseline (Table III). At Week 12, IXE Q2W resulted in complete resolution in a significantly greater percentage of patients than PBO in the scalp, nails, face, and pubis, as well as psoriasis affecting intertriginous regions including the axillae, inframammary fold, inguinal creases, gluteal cleft, and perianal region. Statistically significant improvements versus PBO (p < 0.001) were observed as early as Week 4 (the first post-baseline visit assessed) for all regions except the axillae, which was significant at the second assessment at Week 12. These rates of resolution persisted through Week 52, and response rates at Week 52 were consistent between the 2 treatment groups.
Table III. Complete resolution of psoriasis through 52 weeks of treatment by body region
Safety outcomes during the 12-week blinded treatment period of IXORA-Q have been previously reported (10). In the all IXE exposures population (n = 140), treatment-emergent adverse events (TEAEs) were reported by 107 (76%) patients, and most were mild or moderate in severity (Table IV). Serious AEs were reported by 8 (5.7%) patients and there were no deaths during the study. Three (2.1%) patients receiving IXE discontinued due to AEs of impetigo, bronchitis, and eczema.
Table IV. Adverse events in all patients treated with ixekizumab (IXE) through 52 weeks of treatment in IXORA-Q
Treatment-emergent infections were reported by 75 (53.6%) patients in the all IXE exposures population; most were mild to moderate in severity. Three serious infections occurred, including one Escherichia bacteremia (Escherichia Coli bacteremia), one sepsis, and one cellulitis. The patient who had a serious AE of sepsis also experienced a mild TEAE of diarrhea and a serious AE of colitis that was adjudicated as probable Crohn’s disease. This patient also had a stool sample that tested positive for Campylobacter. The patient discontinued study drug and withdrew from the study, recovered from sepsis, and was recovering from colitis.
Two patients discontinued due to infections during the open-label treatment period, including one patient with bronchitis and one patient with impetigo (the same patient who had a serious infection of cellulitis). Treatment-emergent Candida infections occurred in 7 patients, reported as vulvovaginal candidiasis (n = 1), oral candidiasis (n = 3), skin candida (n = 2), and genital candidiasis (n = 1); vulvovaginal mycotic infection was also reported in 1 of the patients with oral candidiasis. All Candida infections resolved and there were no Candida infections during the blinded treatment period.
Treatment-emergent allergic or hypersensitivity reactions were reported by 14 (10%) patients in the all IXE exposures population; none were anaphylaxis. Injection site reactions were reported by 23 (16.4%) patients in the all IXE exposures population; all were either mild or moderate in severity, none were serious, and none resulted in discontinuation. One treatment-emergent cytopenia (Grade 2 neutropenia) occurred. Two treatment-emergent cerebrocardiovascular events in the all IXE exposures population were adjudicated as peripheral ischemia and atrial fibrillation; neither was categorized as a serious AE or major cerebrocardiovascular event and did not result in discontinuation from the study. Three malignancies (basal cell carcinomas) were reported from the all IXE exposures population during the open-label treatment period; none were serious AEs and did not result in study discontinuation.
Five treatment-emergent events of depression occurred in the all IXE exposures population, none of which resulted in study discontinuation. All were mild or moderate in severity except for one severe event of attempted suicide, which was categorized as a serious AE. The patient who attempted suicide had bipolar disorder (receiving treatment with lithium and desvenlafaxine) and insomnia (receiving treatment with quetiapine), and had a history of suicidal ideation and depression. The suicide attempt occurred following a relationship breakdown and alcohol intoxication, and after missing doses of lithium and quetiapine, with no psychotropic medication taken for 2 days prior to the event. The patient was hospitalized and recovered, resuming treatment with IXE and all other medications. The suicide attempt was determined by the investigator to be unrelated to the study drug.
During the blinded treatment period, no patients receiving IXE were in the severe or very severe categories of the QIDS-SR16. During the open-label treatment period, one patient was in the severe category at Weeks 24 and 40, and no patients were in the very severe category (Table SIII). As measured by the C-SSRS, 11 patients (IXE Q2W: n = 3; PBO: n = 8) had a history of suicidal ideation and/or behavior prior to baseline, 3 of whom reported suicidal ideation or behavior post-baseline. Of these 3 patients, the suicidal ideation or behavior occurred during the open-label treatment period for 2 patients randomized to IXE (including the one attempted suicide discussed above) and one in the PBO/IXE Q4W population. Among patients with no prior history of suicidal ideation or behavior, 3 patients reported suicidal ideation and/or behavior post-baseline, including 2 patients during the blinded treatment period (both receiving PBO) and 1 patient during the open-label treatment period (initially randomized to IXE).
Of all ADA-evaluable patients (n = 139), 15 (10.8%) had TE-ADAs during at least 1 study visit. The majority (n = 10, 66.7%) of TE-ADA-positive patients were classified as low titer (titer < 1:160), 5 were classified as moderate titer (titer ≥ 1:160 and < 1:1280), and no patients were classified as high titer (titer ≥ 1:1280). One (0.7%) patient was positive for neutralizing TE-ADAs at Week 24 only. Of 139 ADA evaluable patients, 23 (16.5%) had injection site reactions, 19 (82.6%) of whom did not have TE-ADAs. Allergic/hypersensitivity reactions were reported by 14 patients (10.1%), 12 (85.7%) of whom did not have TE-ADAs. Injection site reactions were reported by 4 (26.7%) TE-ADA positive patients and allergic/hypersensitivity reactions (no anaphylaxis) were reported by 2 (13.3%) TE-ADA-positive patients.
In the current study, IXE treatment resulted in rapid and persistent clinical improvements in genital psoriasis for up to one year of treatment as measured by both clinician-reported (sPGA-G) and patient-reported (PatGA-Genital) outcomes. Genital psoriasis improvements were consistent between patient subgroups with lower (1% to < 10%) and higher (≥ 10%) BSA involvement. Complete clearance of genital skin was achieved by up to 60% of patients at Week 52. Similarly, among patients with psoriasis affecting sensitive or visible body regions such as the gluteal cleft and face, the majority of patients achieved complete resolution of psoriasis in these regions at Week 52. Rapid and persistent improvements with IXE were observed in overall psoriasis, genital itch, the impact of genital psoriasis on frequency of sexual activity, as well as in pain and other bothersome signs and symptoms of genital psoriasis (15). Clinical responses were generally consistent between treatment arms in the open-label period.
The majority of TEAEs were mild or moderate in severity and no deaths occurred. Serious infections were reported by 3 (2.2%) patients, and 2 (1.4%) patients discontinued due to treatment-emergent infections. There were 7 (5%) treatment-emergent Candida infections reported in IXE-treated patients; none occurred during the blinded treatment period. There were no grade 3 or 4 cytopenias, severe injection site reactions, or discontinuations due to injection site reactions, and no cases of anaphylaxis. One serious AE of colitis occurred in a patient who tested positive for Campylobacter and was adjudicated as probable Crohn’s disease; the patient discontinued study drug, withdrew from the study, and was recovering from colitis at the time of the last visit. One suicide attempt occurred in a patient with bipolar disorder and a history of suicidal ideation and depression. The suicide attempt was determined to be unrelated to study drug by the investigator and the patient continued treatment with IXE.
Although genital psoriasis is a common manifestation of plaque psoriasis associated with reduced quality of life and sexual health (2, 3, 7), it is frequently overlooked in clinical practice (7–9), and there have been few clinical studies evaluating therapies for genital psoriasis (4, 14, 16–18). Hence, well-established treatment options for genital psoriasis are limited and awareness among both healthcare providers and patients is limited. Increased awareness, especially among healthcare providers, is of particular importance because patients with genital psoriasis may present to a wide spectrum of medical specialties (6).
Genital psoriasis often results in a high negative impact on health-related quality of life for patients (19). Recent studies suggest that psoriasis affecting the nails, scalp, axilla, and inframammary folds is associated with the presence of genital psoriasis (3). In fact, genital psoriasis may also be accompanied by intertriginous psoriasis, and in some studies has been classified as a form of intertriginous psoriasis. However, genital psoriasis is a distinct clinical manifestation of psoriasis and can occur independently of intertriginous psoriasis (20). In a prospective cross-sectional study of 852 patients with psoriasis, only 22% of patients with genital psoriasis also had associated intertriginous psoriasis (20). Similarly, in a questionnaire-based study of patients with psoriasis, 38% (n = 197) of patients with genital psoriasis (n = 519) indicated they did not have intertriginous involvement. Furthermore, 20% (n = 310) indicated they had intertriginous psoriasis with no current genital involvement, 77% (n = 234) of whom indicated they had never had genital involvement (21).
IXORA-Q is the first study to provide long-term data on the efficacy and safety of IXE for genital psoriasis for up to one year of treatment. This study had a shorter duration than studies of IXE for the treatment of overall plaque psoriasis (22). Additional limitations of the study include a predominantly white and male patient population, the lack of an active comparator (due to a lack of well-established effective treatments for genital psoriasis currently registered), and the absence of a PBO comparator arm following Week 12 of the study. In addition to genital psoriasis, this study assessed the ability of IXE to clear psoriasis in multiple bothersome body regions. Because only complete resolution of baseline psoriasis in these regions was evaluated and presence/absence was not recorded at every visit, quantitative analysis of the improvement in the severity of psoriasis in these body regions was not determined and complete resolution may have occurred earlier than was detected.
IXORA-Q is the only Phase 3, randomized, controlled trial to evaluate the efficacy of any treatment for genital psoriasis. The study included a geographically diverse and clinically relevant population representative of patients who would be candidates for systemic therapy for genital psoriasis. By including patients with as low as 1% BSA involvement, the study evaluated the efficacy of IXE in clinically relevant patients with moderate-to-severe disease affecting lower (1% to < 10%) and higher (≥ 10%) ranges of body surface involvement. An additional strength of IXORA-Q is that it included newly developed and validated clinician- and patient-reported outcome measures to better assess the overall impact of genital psoriasis on patients.
During a 12-week blinded treatment period of IXORA-Q, IXE resulted in rapid and significant improvements compared with PBO in genital psoriasis, the impact of genital psoriasis on sexual frequency, and the symptoms of genital psoriasis (10). These improvements persisted for up to 1 year of treatment. The safety profile of IXE in patients with moderate-to-severe genital psoriasis was consistent with the safety profile in previous studies of patients with moderate-to-severe overall psoriasis.
The authors thank the participants, investigators, and study staff who made this study possible.
This study was sponsored by Eli Lilly and Company. Medical writing assistance was provided by Cassandra Haley, PhD, CMPP of ProScribe – Envision Pharma Group, and was funded by Eli Lilly and Company. ProScribe’s services complied with international guidelines for Good Publication Practice (GPP3).
Eli Lilly and Company was involved in the study design, data collection, data analysis, and preparation of the manuscript.
Conflicts of interest: LG has been a consultant, investigator, and speaker for AbbVie, Amgen, Celgene, Eli Lilly and Company, Janssen, Leo Pharma, Merck, and Pfizer; has been a speaker and consultant for Valeant and Tribute; and has been an investigator for UCB. JW has been an investigator for AbbVie, Amgen, Biogen, Boehringer Ingelheim, Celgene, Eli Lilly and Company, GlaxoSmithKline, Janssen, Leo Pharma, Merck, Novartis, Pfizer, Regeneron, Stiefel, and Valeant; has been a speaker and served on an advisory board for AbbVie, Eli Lilly and Company, and Janssen. YP has received grant funding or honoraria for services as an investigator, speaker, or member of advisory boards from: AbbVie, Amgen, and Janssen, and has received grant funding for services as an investigator for: Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Dermira, Eli Lilly and Company, Galderma, GlaxoSmithKline, Leo Pharma, MedImmune, Merck, Novartis, Pfizer, Serono, and UCB. LS has served on an advisory board for AbbVie, Eli Lilly and Company, Novartis, and Sanofi and has been an investigator for AbbVie, Amgen, Ascend Biopharma, Australian Wool Innovation Limited, Celgene, Dermira, Eli Lilly and Company, Galderma, Genentech, GlaxoSmithKline, Leo Pharma, Merck, Novartis, Otsuka, Pfizer, Phosphagenics, Regeneron, and UCB. CR has been a consultant, advisor, and /or speaker for AbbVie, Boehringer Ingelheim, Dermira, Dr. Reddy’s Laboratories, Eli Lilly and Company, Janssen, Leo Pharma, Medimetriks, Novartis, and UCB. APB, RB, JE, KT, and CCB, are employees and own stock in Eli Lilly and Company.
Data sharing statement. Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the US and EU and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.


 Click to show fullsize
Click to show fullsize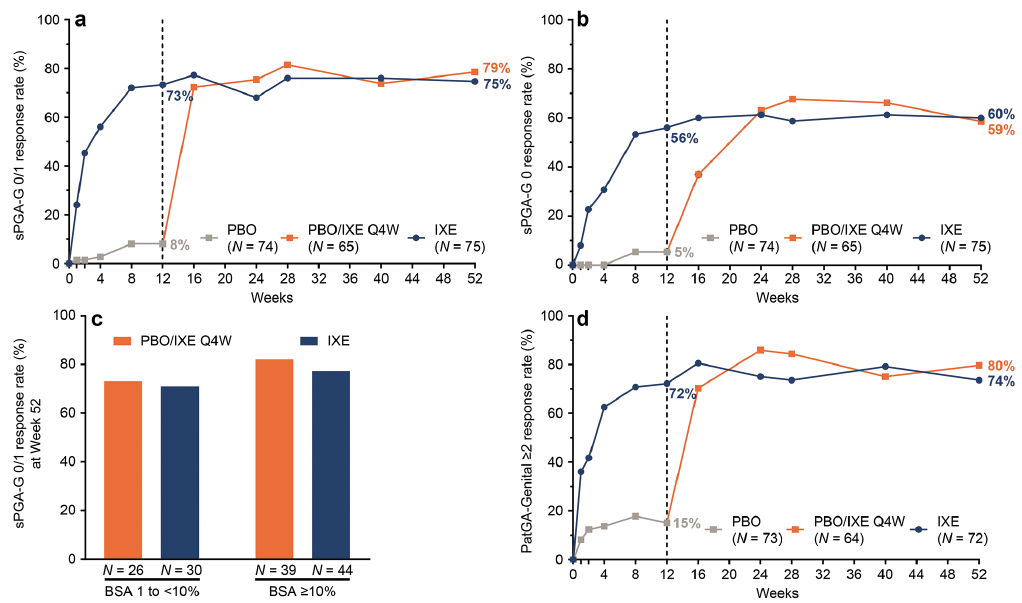 Click to show fullsize
Click to show fullsize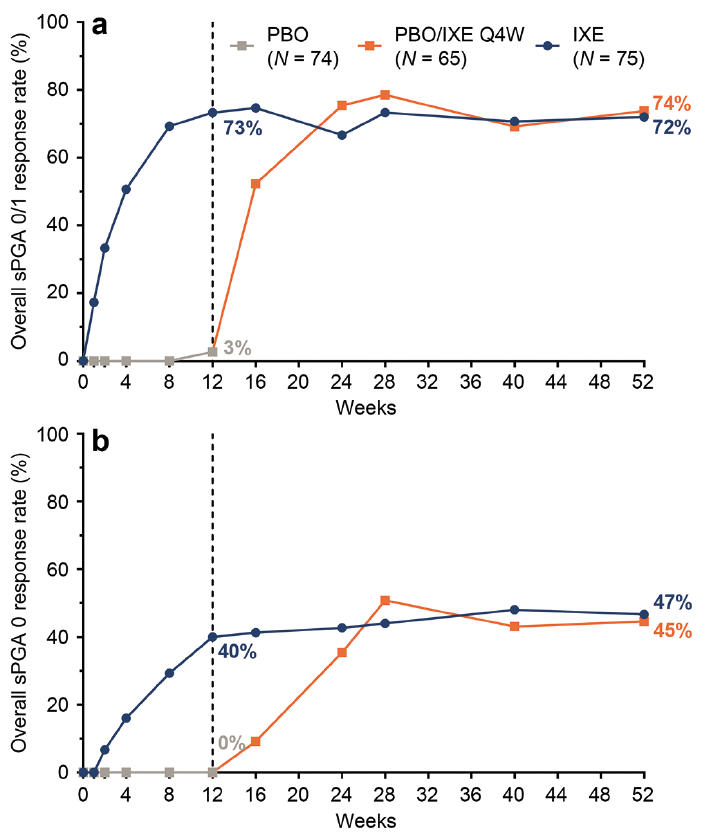 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize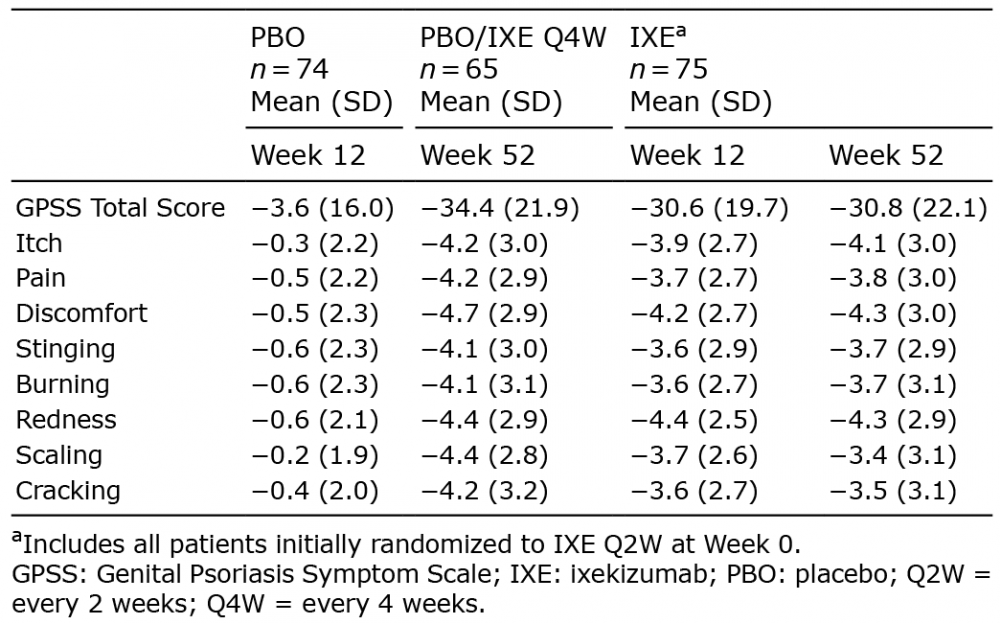 Click to show fullsize
Click to show fullsize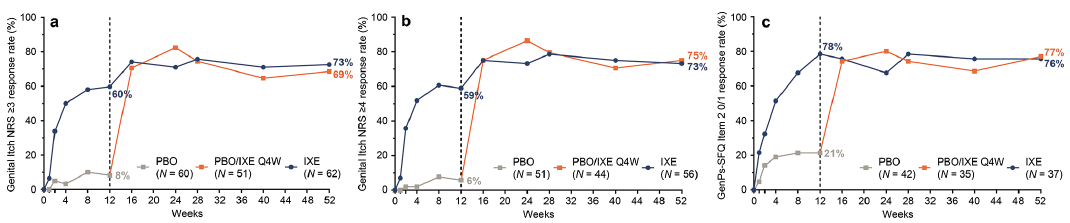 Click to show fullsize
Click to show fullsize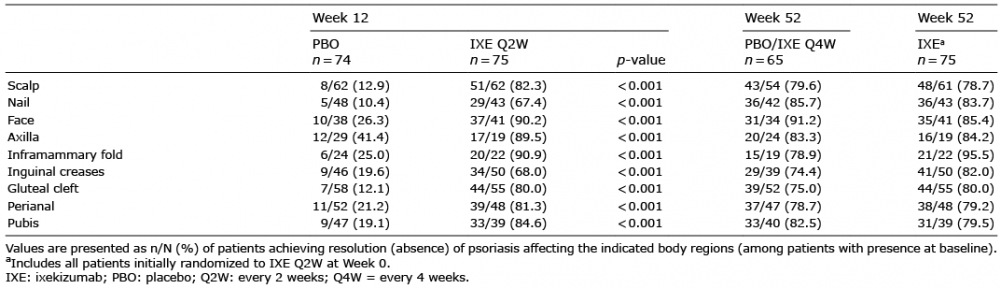 Click to show fullsize
Click to show fullsize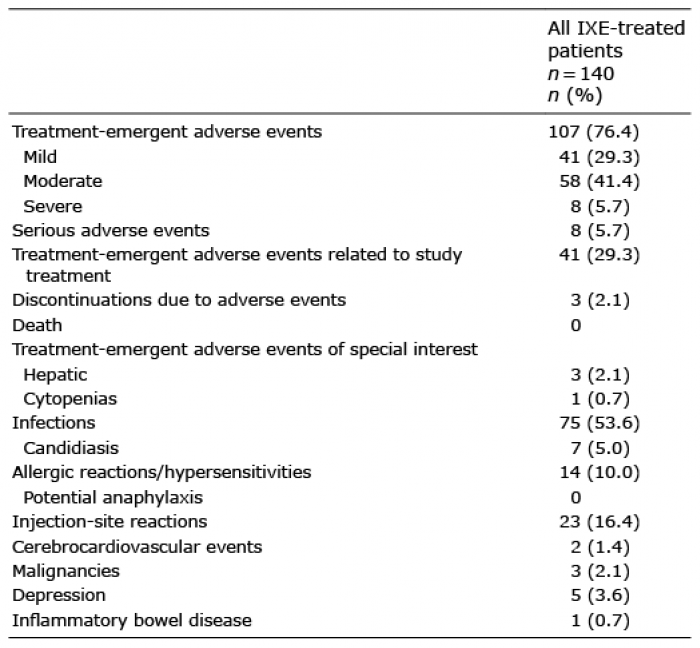 Click to show fullsize
Click to show fullsize