


1Reference Centre for Rare Skin Diseases, Dermatology Department, CHU Toulouse, Paul Sabatier University, Larrey Hospital, FR-31000 Toulouse, 2Epithelial Differentiation and Rheumatoid Autoimmunity Unit (UDEAR), UMR 1056 Inserm – Toulouse 3 University, 3Medical Genetic Department, CHU Toulouse, Toulouse, 4Dermatology Department, CHU Nantes, 5Medical Genetic Department, CHU Nantes, and 6Reference Centre for Rare Skin Diseases, Dermatology Department, CHU Bordeaux, Bordeaux, France. E-mail: mazereeuw-hautier.j@chu-toulouse.fr
Accepted Oct 18, 2019; Epub ahead of print Oct 21, 2019
Acta Derm Venereol 2020; 100: adv00047
Congenital ichthyoses (CI) comprise a heterogeneous group of genetic diseases, which usually present at birth. They affect the entire skin and are characterized by hyperkeratosis and scaling, often associated with skin inflammation. There are many complications including ophthalmic and ear anomalies, pain and pruritus and cutaneous infections. The classification is based on the clinical presentation, and basically distinguishes between non-syndromic ichthyoses (including autosomal recessive congenital ichthyosis (ARCI) and syndromic ichthyoses (1). CI are primarily monogenic diseases with gene mutations leading to a defective skin barrier. To date, ARCI has been associated with a total of 12 genes including SDR9C7 and SULT2B1 for which only a very few families have been reported. We describe here 3 new patients with ARCI caused by mutations in SDR9C7, including 4 previously unreported mutations.
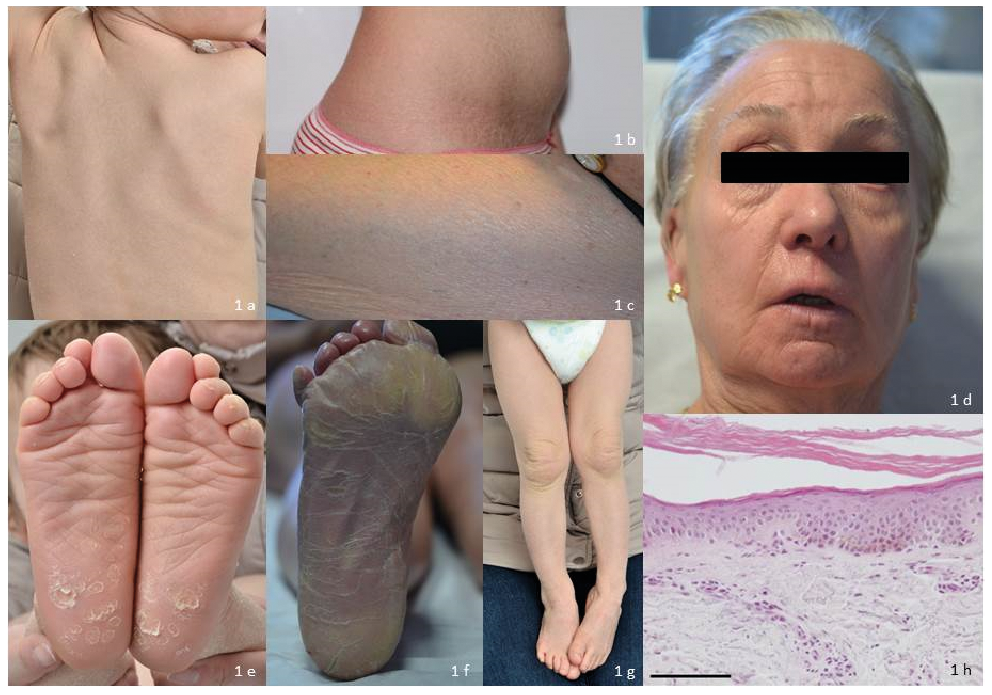
All 3 patients (2 females and 1 male) were born from non-consanguineous unrelated parents of French origin. Their characteristics are reported in Table SI. Only one patient (P1) was born as a collodion baby, and the 2 remaining patients presented with scaly skin with (P2) or without (P3) erythroderma at birth. Intriguingly, past medical history revealed growth delay for P2 and P3, together with puberty delay for P2 and speech delay for P3. All had a mild ichthyosis with generalized scaly skin (fine, large, whitish or light-brown scales). P3 had a ribbed appearance with a mild erythema along the ribs (Fig. 1c). Only one patient had a sparing of the face. P1 has had an ectropion of moderate severity since the age of 40 years (Fig. 1d). All 3 patients had a mild palmoplantar keratoderma (PPK) (Fig. 1e, f) but only 2 patients had hyperkeratosis affecting the elbows and knees (Fig. 1g). None had alopecia, significant hypohidrosis or fungal infections.
Histological examination of a skin biopsy (P1) was reminiscent of lamellar ichthyosis (LI) and revealed marked orthohyperkeratosis, normal granular layer, absence of acanthosis or inflammatory cell infiltration (Fig. 1h).
Informed consent was obtained from the patient and their parents, and the current study was performed according to the principles of the Declaration of Helsinki. Mutation screening of genomic DNA from the 3 patients was performed by next generation sequencing, as described previously (2). Among the 32 genes involved in ichthyoses screened with our AmpliSeq Custom Panel, no potentially pathogenic variations were identified except in SDR9C7, which were verified by bidirectional Sanger sequencing of the corresponding exons. These mutations and the mutations reported previously in the literature are shown in Table SII.
Fig. 1. Clinical and histological character-istics of the 3 patients: mild ichthyosis with generalized scaly skin ((a) fine, large, whitish (P3) or (b) light-brown scales (P2), (c) ribbed appearance with a mild erythema along the ribs (P1), (d) ectropion of moderate severity (P1), (e) mild (P3) or (f) moderate palmoplantar keratoderma (P1), (g) hyperkeratosis affecting knees (P3)). (h) Haematoxylin and eosin staining of affected patient P1’s skin section (inner arm) shows hyperkeratosis, normal granular layer and no inflammatory cell infiltration (bar=100 µm). Permission given to publish these photos.
The novel homozygous missense mutation c.704G>A (p.Gly235Glu, rs756591404) in P1 was already referenced in some databases, but showed a very low frequency in the Exome Aggregation Consortium (ExAC) with no homozygous individuals for the alternate allele. It affected a highly conserved position and the substitution Gly235Glu was predicted to be deleterious for the protein (in silico protein prediction tools SIFT, Polyphen-2).
P2 was compound heterozygous for c.551A>G (p.Asp184Gly) and c.563G>A (p.Arg188His). The mutation c.551A>G had already been reported in 2 families and may affect the function of the protein (3, 4). The mutation c.563G>A was unreported. It was predicted as disease-causing by disturbing normal splicing. Interestingly, a similar pathogenic mechanism was predicted for the previously reported c.562C>T (p.Arg188Cys) mutation, which affected the same codon (3).
P3 was compound heterozygous for c.309A>G (p.Trp103Ter) and c.826C>T (p.Arg276Cys). The nonsense variant c.309A>G was unreported and not referenced in databases. It can be predicted to result in either mRNA decay or in the synthesis of a truncated protein. The previously unreported mutation c.826C>T showed a very low frequency with no homozygous individuals for the alternate allele and was predicted to be deleterious for the protein (in silico protein prediction tools SIFT, Polyphen-2).
SDR9C7 was previously shown to be abundantly expressed in the granular and the cornified layers of the epidermis (5). It belongs to the protein superfamily of short-chain dehydrogenases/reductases (SDRs), known to catalyse activation/inactivation of prostaglandins, retinoids and several classes of steroid hormones, and could be involved in vitamin A metabolism (6). However, the biological function of the protein in the epidermis and its involvement in the pathophysiology of ARCI caused by SDR9C7 mutations remain unclear.
The literature reports 17 other families of various geographic origins with ARCI associated with SDR9C7 mutations (3–5, 7–12). The data from all these patients, summarized in Table SI, help to better define the phenotype of ARCI associated with SDR9C7. Similarly to other genes of ARCI, there is no strict genotype-phenotype correlation but some clinical characteristics may be underlined. All but one started at birth and a collodion baby seems to be rare. Furthermore, the mild degree of erythema (with a peculiar ribbed aspect along the ribs in some patients (Fig. 1c)), the absence of alopecia and the rarity of the ectropion are also interesting findings. Some clinical features appear to be suggestive of this diagnosis, even if inconstantly seen, such as hyperkeratosis and recurrent fungal infections (rarely reported in other forms of CI (13)).
The patients’ conditions evolved into a mild form of LI, except for one family (7), who presented a more severe form. There is also a possibility of skin improvement over time (reported for some families). This may have an impact on the variability of the severity of the phenotype, together with the nature of the mutations, the influence of the environment or some modifying genes.
In conclusion, these new cases expand the knowledge of the molecular bases of ARCI and help to better define the patient’s characteristics and the prognosis of this form of ichthyosis. New cases will make it possible to better define the exact frequency of this ARCI-associated gene.
Conflicts of interest. JM-H is a consultant for ARROW. The other authors have no conflicts of interests to declare.


 Click to show fullsize
Click to show fullsize