


Psoriasis is a common inflammatory skin disease caused by the interplay between multiple genetic and environmental risk factors. This review summarises recent progress in elucidating the genetic basis of psoriasis, particularly through large genome-wide association studies. We illustrate the power of genetic analyses for disease stratification. Psoriasis can be stratified by phenotype (common plaque versus rare pustular variants), or by outcome (prognosis, comorbidities, response to treatment); recent progress has been made in delineating the genetic contribution in each of these areas. We also highlight how genetic data can directly inform the development of effective psoriasis treatments.
Key words: psoriasis; genetics; precision medicine; disease progression; treatment outcome.
Accepted Nov 28; 2019; Epub ahead of print Jan 23, 2020
Acta Derm Venereol 2020; 100: adv00030.
Corr: Jonathan N. Barker, St John’s Institute of Dermatology, King’s College London, 9th Floor Tower Wing, Guy’s Hospital, London SE1 9RT, UK. E-mail: jonathan.barker@kcl.ac.uk.
Psoriasis has benefited greatly among dermatological conditions from genome-wide association studies (GWAS) of increasingly large, clinically well-described samples. Sixty-five regions of the genome have been linked to psoriasis risk in Europeans, with the largest contribution due to HLA-C*06:02, a variant of an important gene involved in immunity. Other regions implicate numerous immune and skin barrier processes in psoriasis development. Recent GWAS-based research has shown that genetics can help distinguish subgroups of psoriasis patients characterised by type (pustular vs. plaque psoriasis), development of joint disease or response to various drugs. This may help inform future tailored treatment strategies for individuals with psoriasis.
Dermatological research has made extraordinary progress over the past 100 years. This has been matched – if not exceeded – by advances in the field of genetics, particularly in the two decades since the initial mapping of the human genome (1, 2). Recent insights into the genetic basis of the common skin disease psoriasis illuminate the translational potential of genetic studies, having directly informed the design of several powerful biologic therapies and small molecule inhibitors.
Psoriasis is a chronic immune-mediated inflammatory disease that affects around 2% of the world’s population (3). It has been designated a serious non-communicable disease by the World Health Organisation and its increasing prevalence represents a substantial global public health burden (4). Genetic research has delivered critical insights into the biology of psoriasis. We now know that psoriasis is a multifactorial disease caused by the interplay between multiple inherited alleles (Box 1) and environmental risk factors. Indeed, it has a particularly strong genetic component among complex diseases, with heritability estimated to exceed 60% (5).
Unlike other biological features, the genome is fixed at birth and does not vary by cell or tissue type, or in response to stimuli: in this sense it reveals the causal biology of psoriasis. In this review, we describe how genetic studies have helped to disentangle pathogenic mechanisms of psoriasis and informed the selection of therapeutic targets. We also highlight the potential of genetic biomarkers as a stratification tool for the effective clinical management of psoriasis.
Early genetic findings
It has long been observed that the incidence of psoriasis is significantly higher among first- and second-degree relatives of sufferers than the general population (6, 7), and it is more concordant among monozygotic than dizygotic twins (8–10).
Linkage studies identified at least 9 genomic regions (loci) that co-segregated with psoriasis (PSORS1-9) in multiplex pedigrees. However, most of these findings could not be replicated, which underscores the limitations of linkage approaches for the analysis of multifactorial conditions (11). A notable exception is the PSORS1 region, which maps to the class I interval of the major histocompatibility complex (MHC) that primarily encodes genes involved in antigen presentation (12–14). The region also contains the candidate gene corneodesmosin (CDSN), which encodes a desmosomal protein involved in keratinocyte cohesion and desquamation (15). PSORS1 has the largest effect size and accounts for 35–50% of disease heritability explained by known loci. Despite the complex correlation structure across the MHC due to extensive linkage disequilibrium (Box 1) (16), HLA-C*06:02 is now confidently considered the most likely causal susceptibility allele, since single nucleotide polymorphisms (SNPs; Box 1) that tag this allele have generated the most significant association signals in subsequent case-control studies (17, 18). Fine mapping studies have suggested the presence of additional association signals within PSORS1, some of which are population-specific (19–22).
The only other successfully validated linkage results are the PSORS2 and PSORS4 loci on chromosomes 17q25 and 1q21, respectively. The most likely susceptibility gene in PSORS2 is CARD14, which encodes a nuclear factor-κB (NF-κB) activator and harbours variants associated with rare and common forms of psoriasis (23–25). PSORS4 contains the late cornified envelope (LCE) genes, which encode stratum corneum proteins involved in terminal epidermal differentiation. This locus has been implicated in psoriasis susceptibility in genome wide association studies of both European and Chinese populations (26, 27).
Psoriasis in the GWAS and post-GWAS era
Genome-wide association studies (GWAS) use highly optimised microarrays that can efficiently and robustly genotype several million genetic markers across the genome. With sufficiently large sample numbers, GWAS allows even small differences in allele frequencies between disease cases and unaffected controls to be detected, making it a much more powerful approach than linkage analysis. As such, GWAS have fundamentally changed the genetic dissection of common complex diseases such as psoriasis. By 2010, initial GWAS efforts in psoriasis had identified 21 susceptibility loci in Europeans (17, 18, 28, 29).
One inherent limitation of GWAS, however, is that it only uncovers statistical relationships. The genetic variants identified by GWAS may actually, by virtue of linkage disequilibrium, be tagging a separate ‘causal’ variant that exerts a biological effect and modifies disease risk. To refine GWAS signals and thus identify potential causal susceptibility alleles, genotyping arrays with dense coverage in regions of interest have been employed. The immunochip included 200,000 SNPs focused in known susceptibility loci for a range of immune-mediated diseases (30). In psoriasis, meta-analysis of immunochip data almost doubled the number of known susceptibility loci and uncovered candidate causal variants at 10 loci including in the innate immunity genes DDX58 and CARD14 (31).
More recently the exome chip aimed to comprehensively genotype protein-altering variants, including rare variants. Exome chip meta-analysis of 12,000 psoriasis cases and 29,000 controls highlighted potential functional SNPs within 11 known psoriasis susceptibility loci. This study provided novel insights into the complex role in psoriasis susceptibility of rare variants in the type I interferon signalling genes IFIH1 and TYK2 (32).
Rather than physically genotyping additional SNPs that are not included in GWAS arrays, however, it is becoming standard practice to perform genome-wide imputation (Box 1) using freely-available computational resources (33, 34). Imputation has been critical in facilitating the larger psoriasis meta-analyses, which combine data generated by different GWAS platforms (35–37). Indeed, an improved imputation strategy revealed a novel psoriasis susceptibility locus at DLEU1, linked to apoptosis, in previously analysed GWAS data (37).
Finally, combining datasets from international collaborations in meta-analyses of genome wide association studies has been essential to enhance statistical power and uncover novel disease susceptibility loci (18, 28, 29, 31, 38). A recent meta-analysis of psoriasis GWAS with a combined effective sample size of > 39,000 individuals identified 16 novel disease-associated regions (36).
As a result of GWAS, targeted association and meta-analysis efforts, the number of independent genomic loci contributing to susceptibility to common plaque psoriasis in populations of European ancestry now stands at 65 (32, 36, 37). More than 30 loci have been implicated in Han Chinese individuals (39). Although these susceptibility loci can span many genes, many of the lead SNPs lie in proximity to genes involved in specific adaptive and innate immune pathways. These include genes involved in antigen presentation (HLA-C, ERAP1), T17 cell activation (IL23R, IL23A, IL12B, TRAF3IP2), innate antiviral immunity/type I interferon signalling (RNF114, IFIH1) and skin barrier function (LCE3B/3D) (Fig. 1) (17, 18, 26, 28, 40–42). The coding variants in genes such as IL23R, TYK2 and TNFSF15 uncovered by targeted association analyses further underscore the involvement of the interleukin (IL)-23/T17 axis in disease pathogenesis (31, 32, 38, 39, 43).
Genetic studies have thus provided important mechanistic insights into the aetiology of psoriasis, and support a pathogenic interplay between immune activation and disruption of skin barrier function (44). There is also evidence of gene-gene interactions (epistasis) contributing to disease heritability, since variants in ERAP1 (encoding an enzyme that trims peptide antigens for loading onto MHC class I molecules) only confer disease susceptibility in individuals also harbouring the HLA-C risk allele (18). Once GWAS association summary statistics are in hand, there are several additional in silico approaches that can help to pinpoint relevant causal genes and variants before costly hypothesis-driven functional experiments are undertaken.
Statistical fine-mapping jointly considers correlated groups of associated variants to estimate the likely causality of each (45). This has been undertaken for several psoriasis susceptibility loci, revealing multiple independent association signals (46).
Pathway analysis methods look for known biological pathways for which gene annotations are enriched across multiple susceptibility loci. NF-κB and type I interferon signalling pathways have thus been implicated in psoriasis pathogenesis (36).
If GWAS summary results are available from other studies that have assessed the genetic basis of relevant molecular traits, colocalisation with the disease association signal can be assessed (47, 48). In particular, expression quantitative trait loci (eQTLs) are SNPs associated with the level of expression of a gene in a specific tissue. Colocalisation of a psoriasis susceptibility signal and a skin- or immune-based eQTL would thus provide strong evidence that the variant directly modifies psoriasis risk and suggest a probable mechanism of action. This powerful approach has been successfully employed in GWAS studies of acne (49) and atopic dermatitis (50) but has yet to be employed systematically in a large psoriasis dataset, with only suggestive colocalisations being reported in cross-disease studies (51, 52).
It is worth remarking that all of these approaches rely to a greater or lesser extent on open science: the continuing efforts of research groups around the world that are committed to making reference data, summary results, annotations, tools and computational resources publicly available in the interests of collaborative science.
Fig. 1. Biological pathways implicated in psoriasis pathogenesis via genome wide association studies (GWAS). Candidate causal genes from selected disease-associated loci identified by GWAS. Arrows signify the crosstalk between the immune pathways shown (e.g. interleukin (IL)-17 and type I interferon signalling both activate nuclear factor-κB (NF-κB) pathways). Red boxes: genes involved in mechanisms currently targeted by psoriasis treatments.
The genetic insights gained from large-scale association analyses have paved the way for transformative novel therapeutics in psoriasis. Indeed, it has been shown in general that pipeline drugs whose mechanisms are supported by direct genetic evidence are more likely to reach the clinic (53, 54). Based on the mechanistic insights that have emerged from genetic studies in psoriasis, the IL-23/T17 axis has been a particular focus for drug development. Biologic agents such as ustekinumab (targeting the common p40 subunit of IL-12 and IL-23), secukinumab and ixekizumab (targeting IL-17A), and newer monoclonal antibodies targeting the p19 subunit specific to IL-23 (including guselkumab and tildrakizumab), have shown progressively increasing efficacy rates in clinical trials (55). These agents are now licensed for use in the USA and Europe and have impressive effectiveness and tolerability in real world practice (55, 56).
In addition to informing the targets of biologic medications, genetic studies have opened new avenues for small molecule therapeutics. Following genetic association data highlighting TYK2 as a causal allele (31, 32), an oral, selective inhibitor was developed, which has shown promising efficacy in phase II trials (57).
Missing heritability
Despite the recent progress in psoriasis genetics, less than a quarter of heritability is thought to be explained by the susceptibility loci identified to date (32). There are several reasons for this missing heritability.
A substantial fraction of heritability may arise from rare variants that are not geno-typed or well tagged by GWAS arrays. A recent analysis of 22,000 whole genomes makes a compelling case for this, since the heritability of both height and body mass index could be almost fully explained when very rare variants are accounted for (suggesting also that pedigree-based estimates of heritability are not overestimated) (58). The same may be true for psoriasis susceptibility, although sequencing efforts will need to surpass those performed to date (59) to confirm this.
More generally, the estimated heritability explained at a GWAS-identified locus may be underestimated where the lead GWAS SNP is a poor tag for the true causal variant, or where there are multiple true causal variants (60).
Another explanation could be high polygenicity, where many common SNPs across the genome may modify psoriasis risk, but with effect sizes too small to have been identified with current GWAS sample sizes. Although increasingly large case-control study populations will help to address this (61), sufficient numbers to fully elucidate the role in psoriasis pathogenesis of every individual common SNP are impractical. One approach to overcoming this limitation is to consider genetic variation aggregated according to known biological function. For example, functional network-based analyses have been applied to suggest novel mechanisms involved in psoriasis (36, 62).
It could also be the case that psoriasis risk attributable to individual genetic variants does not accumulate additively and independently, so that simple GWAS association tests mask more complex causal biology. Alternative models of genetic architecture have been explored (63), including genetic interactions genome-wide (64, 65) (recall the HLA-C/ERAP1 interaction described previously).
Missing heritability in genetic studies could be due in part to epigenetic variation: DNA modifications that can cause differences in gene expression even when no differences are present in DNA sequence. Numerous studies have begun to explore the role of epigenetics in psoriasis, although the types of modification and study designs have varied widely, making it difficult to assess their overall contribution to heritability (66).
The complex genetic nature of psoriasis and the unresolved missing heritability have implications for the growing industry of direct-to-consumer genetic testing. While genetic risk profiles can offer additional information beyond family-history based risk estimates (67), this information will likely be insufficiently precise or consistent to offer substantial clinical utility (68, 69) and it is vulnerable to misunderstanding by the public (70, 71).
As we shall describe, however, the genetic risk profiles of larger cohorts still hold great potential to refine our understanding of the biology and to inform effective clinical management of psoriasis.
The possibilities of GWAS-based analysis have now moved beyond the study of simple susceptibility and towards disease stratification. With large collections of genotyped and deeply phenotyped individuals, the genetic basis of many other aspects of psoriasis natural history and treatment response can be characterised (Fig. 2). These collections could comprise psoriasis patients (e.g. PSORT (72)) or be derived from the general population with phenotype data from linked electronic medical records (e.g. UK Biobank (73)).
These “post-susceptibility” genetic studies still currently utilise much smaller samples than the susceptibility GWAS meta-analyses described above. However, they can benefit from numerous methods that incorporate or compare genetic information (typically GWAS summary statistics) from related traits to make novel inferences. Relevant methods include polygenic risk scores (PRS) (74) and genetic correlation (75, 76) to assess shared genetic associations, Mendelian randomisation (77) to assess causality, and methods for deconvoluting genome-wide association signals into functionally relevant constituents (78) (Box 1). While findings from psoriasis susceptibility studies offer a natural starting point (and efforts to accurately document and annotate these associations are ongoing (79, 80)), the utility of these methods are greatly enhanced by the availability of GWAS summary results for thousands of other traits, including physiological, disease-based and molecular traits (81).
We offer here a brief overview of recent progress in psoriasis genetics beyond susceptibility.
Fig. 2. Psoriasis genetics beyond susceptibility. Various strategies are employed to study the genetic factors that influence the conceptual trajectory from risk of psoriasis through disease onset and prognosis to patient outcomes (red arrow). Susceptibility: allele frequencies are compared between psoriasis cases and controls to reveal genetic variants contributing to psoriasis risk. Onset: gene × environment studies may integrate genetic data with environmental exposures (indicated by globe symbol) to identify relationships between genes and environmental triggers; age-of-onset is also influenced by genetic factors and this can be investigated where age-of-onset data (denoted by birthday cake symbol) are available for psoriasis cases. Severity: genetic profiles can be compared between psoriasis patients with mild and severe disease; severity may also be studied as a continuous outcome. Comorbidities: genetic profiles are compared between psoriasis patients with and without comorbid disease D; more sophisticated methods will also consider the genetic basis of disease D in the wider population. Response to treatment: genetic profiles can be compared between psoriasis patients responding and not responding to a treatment; response may also be studied as a continuous outcome. Gen. corr.: genetic correlation; MR: Mendelian randomisation; PRS: polygenic risk score.
Onset
It remains unclear how genetic susceptibility variants interact with environmental risk factors such as infection, ultraviolet exposure, smoking, alcohol, and psychological stress to trigger psoriasis onset (82). Initial findings suggest that the risk attributable to HLA-C*06:02 may be modified by smoking and stress (83). The pathogenic contribution of smoking may also be mediated via variants in CYP1A1, a key gene in the aryl hydrocarbon receptor signalling pathway (84). The availability of large datasets with environmental exposure and GWAS data (such as UK Biobank (73)) now offers the opportunity to study gene-environment interactions in a more systematic manner, and this is an active area of research in the field. Age of onset is better studied. It is well established that the HLA-C*06:02 susceptibility allele is associated with earlier disease onset (85, 86). Tsoi et al. used PRS analysis to show that a greater burden of psoriasis susceptibility variants is associated with earlier disease onset, even when only non-HLA susceptibility loci are considered (36).
Comorbidities
Psoriatic arthritis (PsA), with prevalence estimates ranging from 6–41% among individuals with psoriasis (87), has been the subject of large genetic studies as a disease in its own right (88–90). Particularly revealing, however, are studies comparing individuals with psoriasis and PsA against cutaneous-only psoriasis cases, (either directly or with reference to unaffected controls). Several studies focusing on the HLA region suggest that certain HLA-B alleles, including HLA-B*27, are associated with increased PsA risk in the presence of psoriasis (21, 91, 92), while HLA-C*06:02 is not (91). Genome-wide analysis has identified additional associations of interest, including independent alleles in known psoriasis susceptibility loci (including at IL23R and TNFAIP3) (93). The translational potential of these approaches was recently explored using a “risk score” of 200 genetic markers that proved predictive of PsA development (area under the receiver operator curve = 0.82) (37). While this finding requires replication and may benefit from phenotype refinement (there are at least five recognised subtypes of PsA (94)), it offers a first step towards prognostic genetic risk profiling.
Obesity and related cardiometabolic traits have also been studied. While a large GWAS-based investigation found the genetic architectures of psoriasis and cardiometabolic traits to be largely distinct (95), an epidemiological association with obesity is well established (96, 97) and twin studies suggest a genetic correlation (98). Based on psoriasis and body mass index (BMI) GWAS data, Mendelian randomisation reveals a causal relationship: higher BMI increases the risk of psoriasis, whereas psoriasis does not have a causal effect on BMI (99, 100). Given the relatively large effect that HLA-C*06:02 exerts on psoriasis risk, it may be interesting to examine the causal role of BMI separately in patients positive and negative for this allele.
In principle, the shared genetic aetiology between psoriasis and any other associated condition can be readily explored at scale via GWAS data. A recent example looked at psoriasis alongside 4 other inflammatory diseases (ankylosing spondylitis, Crohn’s disease, primary sclerosing cholangitis and ulcerative colitis), finding genetic overlap between the conditions that may drive co-occurrence, but with the qualification that patients affected by multiple conditions are likely to be genetically distinct from those with a single disease (101). Shared genetic factors have been found to extend beyond inflammatory disease, such as the positive genetic correlation observed between psoriasis and schizophrenia (102).
Stratified medicine
Genomic information has an exciting role in potential future personalised models of disease prevention and treatment (103). Although highly discriminative genetic prediction for complex diseases such as psoriasis (which are influenced by many genetic factors of modest effect) is unlikely (74), there remains ample opportunity to “stratify” individuals into broader groups according to distinct risk and response profiles, thus leading to more effective and economical care.
Effective deployment of expensive biologic therapies is an area of promise in psoriasis. Patients positive for the HLA-C*06:02 psoriasis susceptibility allele demonstrate better response to ustekinumab than HLA-C*06:02-negative patients, particularly during the initial months of treatment (104, 105). Numerous candidate gene studies (and one small GWAS (106)), have tested for genetic associations with response to TNF antagonists such as etanercept, adalimumab and infliximab, often pooling observations for multiple drugs. Robust associations have until recently been scarce, but we are beginning to see better-powered investigations; a recent Danish study found significant associations with anti-TNF response in several immune genes (107). We recently showed via a comparative approach that HLA-C*06:02 status could inform choice of treatment between adalimumab and ustekinumab, particularly when used in combination with clinical factors. Specifically, we found that HLA-C*06:02-negative patients with psoriatic arthritis were significantly more likely to respond to adalimumab than ustekinumab after 6 months (odds ratio, 5.98; p = 6.89 × 10-5), with no such difference observed in HLA-C*06:02-positive patients (108). This has promising clinical utility.
PRS may also help to define strata relevant to the management of psoriasis. Several studies have explored the predictive ability of PRS in psoriasis susceptibility (36, 109, 110) but the true translational benefits of this approach may lie in identifying and characterising groups of patients with very high or very low PRS scores (74). More research in this area in psoriasis is therefore warranted.
Pustular psoriasis is a rare subtype characterised clinically by the presence of sterile pustules on variably erythematous skin, and histologically by diffuse dermal neutrophilic infiltration (111). It can be classified as either acute generalised (generalised pustular psoriasis (GPP)) or chronic localised disease (palmoplantar pustulosis (PPP) and acrodermatitis continua of Hallopeau (ACH)) (112). Pustular psoriasis has a distinct genetic architecture to plaque psoriasis, underscored by a lack of association with the PSORS1 locus (113). The severity and rarity of the clinical phenotype indicate that pustular psoriasis could be associated with rare alleles of moderate to large effect, which has been supported by the identification of three disease genes (IL36RN, AP1S3 and CARD14) using next-generation sequencing technologies (Box 1).
Linkage studies of consanguineous pedigrees and exome sequencing of unrelated GPP patients identified autosomal recessive loss of function mutations in IL36RN (114, 115). IL36RN encodes the IL-36 receptor antagonist (IL-36Ra), which modulates the activity of the IL-1 family cytokines IL-36α, -β and -γ. The screening of expanded patient resources subsequently identified a spectrum of IL36RN mutations that are distributed
throughout the length of the protein and are associated with pustular psoriasis in a variety of populations (116, 117).
Genotype-phenotype analyses indicate that IL36RN disease alleles are less common in individuals with PPP (frequency 0.03) than GPP (0.19) and ACH (0.16) (116). Although recessive IL36RN alleles are typically observed in patients presenting with a severe clinical phenotype (early-onset GPP characterised by a high risk of systemic involvement) (118), deleterious IL36RN variants have also been associated with localised pustular disease (119). Individuals harbouring a single IL36RN mutation are occasionally affected, and they classically present with disease at a later age, indicating a dose-dependent effect (116, 118). Thus, genotype-phenotype analyses provide evidence for variable penetrance of disease alleles and a potential role for genetic modifiers and environmental factors.
Since IL36RN mutations are only found in a minority (~25%) of pustular psoriasis cases (118), exome sequencing was undertaken to gain a better understanding of the genetic basis of the disease. This uncovered two recurring founder mutations in the AP1S3 gene (120). While these defects were found to account for 12% of pustular psoriasis cases of European descent, no AP1S3 mutations were found in Asian patients. AP1S3 encodes the σ1 subunit of AP-1, an evolutionarily conserved hetero-tetramer that has been implicated in the formation of autophagosomes (specialised vesicles that mediate autophagy). Autophagy is an intracellular degradation pathway for misfolded proteins and damaged organelles (121) and has been shown to regulate cutaneous immune responses (122, 123). AP1S3 mutations may lead to defective autophagy, causing accumulation of p62 (an adaptor protein that mediates NF-kB activation) and upregulation of IL-36 mediated cutaneous inflammation (124). Therefore, mutations in different disease genes converge on the de-regulation of IL-36 signalling in pustular psoriasis, highlighting IL-36 blockade as a promising therapeutic strategy regardless of the specific gene affected.
CARD14 was subsequently confirmed as a third disease gene for GPP (25). CARD14 is highly expressed in keratinocytes and encodes a scaffold protein that, upon oligomerisation, mediates TRAF-2 dependent activation of NF-κB signalling. A deleterious gain-of-function substitution in CARD14 has been associated with GPP in an extended case series and shown to cause spontaneous CARD14 oligomerisation in vitro (25). The same variant was also found in two patients with PPP (125), which provides further evidence for an overlap in the genetic basis of generalised and localised forms of pustular psoriasis. Indeed, gain-of-function CARD14 mutations have been detected in cases of familial plaque psoriasis (23, 24), indicating shared aetiological mechanisms in plaque and pustular subtypes of disease.
There is a substantial unmet need for effective treat-ments for pustular psoriasis (111). The conventional systemic agents used for the treatment of plaque psoriasis are often ineffective in pustular phenotypes and there is a paucity of robust clinical trial data, such that current guidelines are mostly based on isolated case reports (111). However, recent exciting progress in this area shows a clear throughline from genetic discovery to treatment advances. IL-1 blockers are being investigated as potential treatments for pustular psoriasis and a multi-centre double-blind randomised controlled trial of anakinra in PPP is currently underway (http://apricot-trial.com/). In GPP, anakinra has been shown to cause initial rapid clinical improvements in case reports (126, 127), although full disease remission was seldom achieved. This incomplete response supports the notion that IL-1 itself is not the dominant disease driver but participates in positive regulatory feedback loops driven by IL-36 (128).
In vivo and ex vivo research has validated IL-36 signalling as a powerful therapeutic target in psoriasis, and indicates that IL-36 blockade would not substantially compromise host defences (129). A recent phase I proof of concept study of 7 patients demonstrated that blockade of the IL-36 receptor (using a single intravenous dose of a monoclonal antibody) reduced the severity of GPP over a 20-week period (130). The agent was efficacious irrespective of the presence of known causal genetic variants and larger scale clinical trials of IL-36 antagonists in pustular psoriasis are currently underway.
Non-European ethnicities
We have shown that genetics will be instrumental in moving healthcare provision towards stratified, and even personalised, models. However, such progress is dependent on robust genetic associations with disease susceptibility, clinical outcomes and other related traits. The majority of genotyping efforts to date have focused on populations of European, and to a lesser extent Han Chinese, origin, meaning the translational potential of GWAS is largely limited to these groups at present.
A trans-ethnic GWAS meta-analysis of psoriasis susceptibility demonstrated heterogeneous genetic associations between European and Han Chinese populations (20). Other ethnic groups in which smaller GWAS and candidate gene studies have been undertaken include Indian (131), Japanese (132) and Omani Arab (133) populations. We are unaware of genetic studies of psoriasis in people of African descent. While lower prevalence might make psoriasis a smaller population burden among predominantly non-white populations (134) the disease burden for individual psoriasis patients is high, and large-scale genetic studies across ethnic groups are warranted.
Such endeavours will benefit from recent community efforts to generate the necessary supporting resources, including statistical tools for trans-ethnic meta-analysis (135), reference panels for genome-wide (136) and HLA allele (137) imputation, and GWAS summary results for common traits (138).
The future of genetics in psoriasis
As with other complex diseases, we believe that genetics will be at the heart of future success in translational psoriasis research. Increasingly large GWAS studies will improve power to detect genetic variants with small effects on psoriasis risk, refining our understanding of the genetic basis of the disease. This increased resolution should allow more accurate deconvolution of susceptibility associations into functional mechanisms of disease, aided by a growing catalogue of systematically derived and publicly available GWAS datasets for intermediate molecular traits. There is also an increasing awareness in the investigative dermatology community of the importance of precise phenotyping. When combined with genetic data, larger and more detailed clinical datasets will help reveal genetic differences between patients that differ in phenotypic presentation or outcome and therefore inform the development and deployment of effective therapies. Finally, as patients become more likely to undergo GWAS profiling or whole-genome sequencing as part of standard healthcare provision, there will almost certainly be benefits to be derived from PRS or related genome-wide measures. These benefits are unlikely to come from very precise diagnostic or prognostic predictions but rather from prioritising individuals for early screening or closer monitoring, thus making optimal use of clinical resources and reducing the significant disease burden of psoriasis at the population level.
ND is supported by Health Data Research UK (MR/S003126/1), which is funded by the UK Medical Research Council, Engineering and Physical Sciences Research Council, Economic and Social Research Council, Department of Health and Social Care (England), Chief Scientist Office of the Scottish Government Health and Social Care Directorates, Health and Social Care Research and Development Division (Welsh Government), Public Health Agency (Northern Ireland), British Heart Foundation and the Wellcome Trust. We also acknowledge support from the NIHR Biomedical Research Centre at King’s College London/Guy’s and St Thomas’ NHS Foundation Trust.


 Click to show fullsize
Click to show fullsize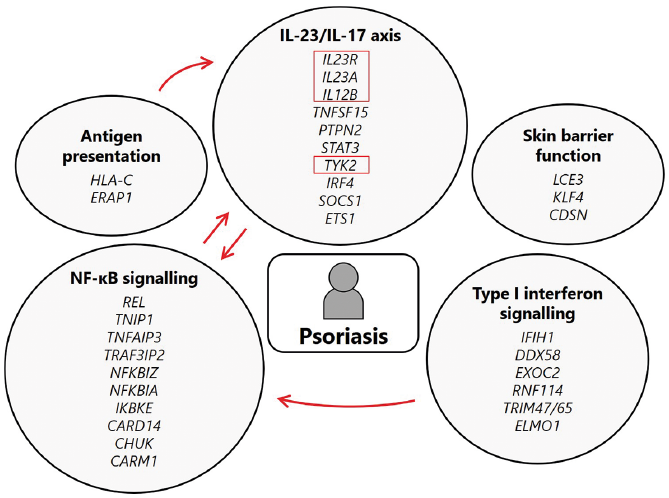 Click to show fullsize
Click to show fullsize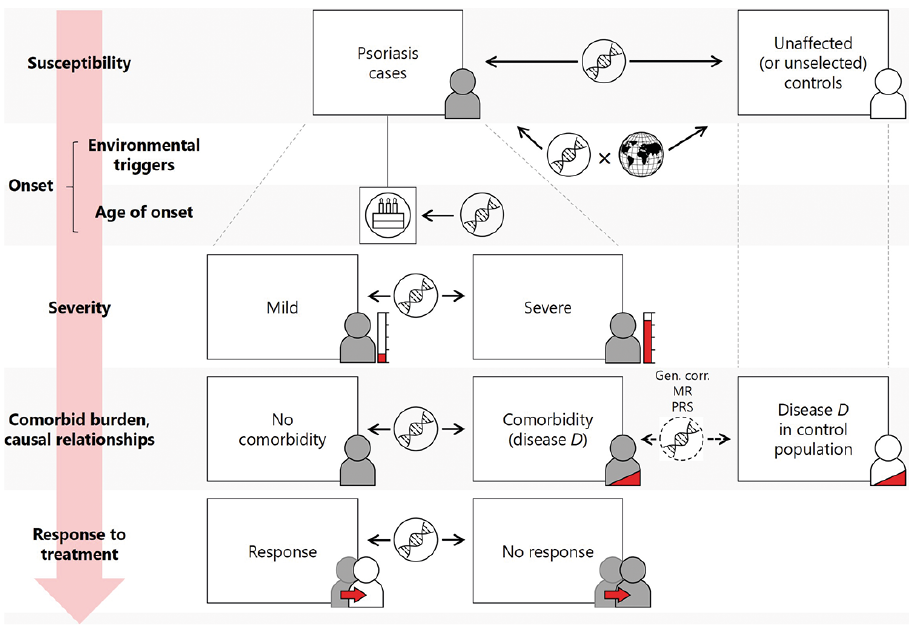 Click to show fullsize
Click to show fullsize