


Department of Dermatology, AP-HP Hôpital Saint-Louis, Université de Paris, Paris, and Laboratory of Genetics of Skin Diseases, UMR INSERM U1163, Institut Imagine, Necker Hospital, Paris, France
Pustular psoriasis is a clinically heterogeneous entity of different, orphan disease subtypes, among which the most clearly defined are generalized pustular psoriasis, palmoplantar psoriasis, and acrodermatitis continua of Hallopeau. Although phenotypically and genetically distinct from psoriasis vulgaris, these subtypes may be associated with plaque psoriasis lesions, establishing the rationale for their inclusion in the psoriasis spectrum. Unlike psoriasis, however, their genetic background is thought to be mainly monogenic, as shown by the recent identification of mutations in 3 different genes of the skin innate immune system; IL36RN, CARD14 and AP1S3. These major advances in the understanding of the disease pathogenesis have led to the design and ongoing development of tailored therapeutic approaches, which are highly necessary given the refractory nature of pustular psoriasis in response to most available antipsoriatic drugs.
Key words: pustular psoriasis; pustulosis; generalized pustular psoriasis; palmoplantar pustulosis; interleukin-36.
Accepted Nov 28; 2019; Epub ahead of print Jan 23, 2020
Acta Derm Venereol 2020; 100: adv00034.
Corr: Hervé Bachelez, Service de Dermatologie, Hôpital Saint-Louis, 1, avenue Claude Vellefaux, FR-75475 Paris cedex 10, France. E-mail: herve.bachelez@aphp.fr
Pustular psoriasis defines a heterogeneous group of skin inflammatory diseases, which have in common the presence of aseptic pustules. Genetically distinct from psoriasis vulgaris, they have been shown to be related to mutations in any of 3 genes of the skin immune system, respectively called IL36RN, CARD14 and AP1S3. These recent advances have initiated the design of biological drugs specifically targeting key actors of inflammation in pustular psoriasis, with interleukin-36 inhibitors as the most advanced example of therapeutic development.
Psoriasis is a chronic inflammatory disease entity that includes different clinical phenotypes, of which the so-called psoriasis vulgaris (PV) or plaque psoriasis variant is by far the most prevalent, representing approxi-mately 80% of cases, and resulting from the combination of a multigenic, complex genetic background with environmental triggers (1, 2). Aside from this most frequent clinical form of psoriasis, much rarer clinical phenotypes, all characterized by the presence of neutrophilic skin inflammation with macroscopically visible, non-infectious or aseptic pustules, have been termed pustular psoriasis and, in some cases, psoriasis-related subphenotypes (2). Long-neglected, these phenotypes have attracted a lot of interest over the last 10 years due to major advances in the understanding of their pathogenic mechanisms, involving a major deregulation of skin innate immune responses, reflected at the histological level by an intense afflux of neutrophils and monocytes in the lesional dermis and epidermis (2). The current review integrates long-established clinical features with the more recently re-defined disease subtypes classification and, more importantly, advances in physiopathological scenarios, which have already driven therapeutic innovations.
Pustular psoriasis consists of several clinical entities, of which the best defined are: (i) localized pustular psoriasis dominated by palmoplantar pustular psoriasis (PPPP), also called palmoplantar pustulosis (PPP); (ii) acrodermatitis continua of Hallopeau (ACH), which predominantly involves acral areas of the hands and/or feet; and (iii) generalized pustular psoriasis (GPP), a disseminated, severe and potentially life-threatening form of psoriasis. The question of whether these pustular skin disorders belong to the psoriasis spectrum has been debated for a long time. Their intersection and overlap with PV is reflected by their frequent coexistence in a given patient, and by some commonalities in their respective mechanistic models. In this sense, studies of pustular psoriasis have also been insightful regarding mechanisms of skin inflammation, both in physiology and for other skin-inflammatory diseases.
Clinical characteristics and diagnostic procedures
GPP, the most severe of all the psoriatic disease variants, is an orphan skin and multisystemic inflammatory disease characterized, in its typical forms, by intermittent flares or attacks with partial or complete remission in between (Fig. 1). Estimated prevalences of GPP are 0.0002% and 0.0007%, in France and Japan, respectively (3, 4). Each flare consists of the acute onset of a rapidly disseminating cutaneous eruption an extensive skin rash covered with aseptic pustules at an early stage, combined at some point with systemic symptoms, such as a variable degree of fever up to 40°C, and general malaise with fatigue. Other extracutaneous manifestations, such as polymyalgia and polyarthralgia, are common, and arthritis may occur (5, 6). Several subtypes of GPP have been defined depending both on disease course and clinical presentation: (i) the acute von Zumbusch type; (ii) GPP in pregnancy, previously named impetigo herpetiformis; (iii) the annular GPP clinical subphenotype; and (iv) GPP associated with PV. GPP is an unpredictable disease; the spectrum of severity of attacks varies widely between patients and in any given patient, ranging from the absence of any systemic symptoms, which does not rule out diagnosis, to the presence of high fever or even life-threatening complications requiring admission to the intensive care unit (6). Biological test abnormalities typically consist of raised serum concentrations of inflammatory proteins, mainly C-reactive protein (CRP), peripheral blood hyperleukocytosis with neutrophilia, and a high prevalence of liver test abnormalities, sometimes delayed with respect to the onset of ongoing GPP flare (7, 8). Extracutaneous manifestations of GPP, such as osteoarthritis, uveitis, acute respiratory distress syndrome, and cardiovascular aseptic shock (the last related to the massive release of inflammatory cytokines), may occur at any stage during the disease course (7, 9–11). The reported high prevalence of liver test abnormalities during GPP attacks, mainly mild to moderate cholestasis and/or cytolysis, raised the hypothesis of aspecific liver/biliary involvement by the inflammatory process. This hypothesis was reinforced by results from magnetic resonance imaging (MRI) of the liver and biliary ducts, showing, in some patients, strictures alternating with dilatations of the principal and/or accessory biliary ducts, and was definitively ascertained by histopathological analysis of liver biopsies showing innate immune cells, mainly neutrophils, infiltrating the epithelium of biliary ducts and the portal and periportal spaces (6). This last entity, which shares features of other types of inflammatory cholangitis, was further termed neutrophilic cholangitis, and seems to have a benign short- and medium-term evolutive profile, although additional follow-up studies are needed to investigate its long-term prognosis (8). More recently, cases of neutrophilic cholangitis have been reported in patients with localized pustular psoriasis, and in those with PV with or without psoriatic arthritis, raising the hypothesis that deregulation of the extracutaneous innate immune system is not exclusive to GPP, but may also be observed in more frequent psoriatic variants (12).
One very specific evolutive feature of GPP is the spontaneously self-remitting pattern of disease flares, at least in classical intermittent forms. Typically, this spontaneous remission of attacks happens in a matter of weeks following the onset of attacks, but there are cases in which chronic skin lesions persist in between attacks of GPP (9). Whatever the genetic background, the intermittent, acutely flaring course of GPP allowed triggering factors to be identified, the best known being infections, stress, corticosteroid treatment withdrawal, and pregnancy (5, 6, 13). Cases of GPP with onset during pregnancy, usually early during the third trimester, have been also termed impetigo herpetiformis, but they are now acknowledged as part of the GPP entity. Their prognosis may be severe both for the mother and the foetus, potentially leading to intrauterine growth restriction, miscarriage, or foetal death (14, 15). Therefore, GPP in pregnancy requires close monitoring of foetal viability. GPP in pregnancy should be considered as a serious, potentially life-threatening situation for both mother and foetus.
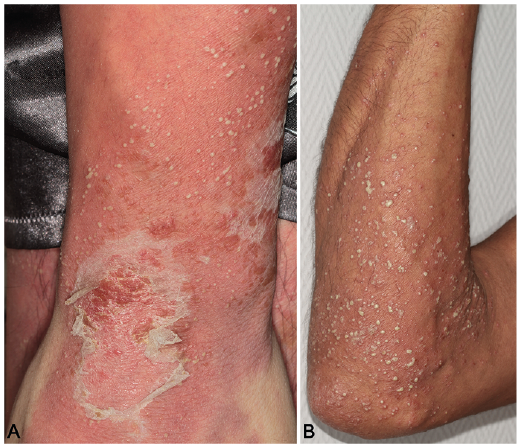
Fig. 1. Skin lesions in patients with generalized pustular psoriasis. (A) A diffuse erythema is covered with confluent pustules, leading to formation of pustular lake, with superficial scaling at a later stage, in a patient free of IL36RN or CARD14 mutation. (B) Disseminated, separated pustules on an erythematous basis of the forearm of an adult patient with identified deficiency of IL36 receptor antagonist.
Physiopathology and prognosis
Infectious respiratory viral triggers have been identified recently by multiplex PCR-based analysis of nasopharyngeal swabs in a small cohort of patients with different subtypes of psoriasis, including GPP (16). Interestingly, viral nucleic acids are potent agonists of the innate immune system, and can stimulate the release of inflammatory cytokines operating in psoriasis pathogenesis, including interleukin-36 (IL-36) (16). The role of these viral triggers has been raised in several studies, and a striking observation is the short time interval between the infection and onset of GPP flare, in keeping with a potent stimulation of the innate immune system (9, 13, 16). The role of these infectious triggers raises the challenge of immune intervention with immunosuppressants during GPP attacks with simultaneous infection, emphasizing the need for effective therapeutic strategies with appealing infectious safety profiles. The deciphering of the pathogenesis due to identification of causal genetic abnormalities, mainly mutations of the IL36RN gene encoding a regulator of the IL-36 inflammatory pathway in a subset of patients with GPP, established a strong rationale for the development of targeted therapies, a crucial breakthrough which is addressed below (13).
Mortality rates for recent cohorts of GPP are not available, but its life-threatening potential is acknowledged. Likewise, some skin and systemic signs and symptoms-based attempts to score the severity of GPP flares have been launched in Japan, paving the way for more specific, reproducible scoring tools, in a disease where PV-specific Psoriasis Area Severity Index (PASI) is not suitable, notably due to the absence of any induration in GPP lesions (7).
Recent advances in the assessment of the severity of pustular psoriatic diseases are addressed below.
PPPP, also called palmoplantar pustulosis (PPP), is the most common of pustular psoriasis variants, and the most common localized pustular variant (Fig. 2) (17). Prevalence estimates of 0.01% have been reported, and the disease predominates in women, with a strong link with smoking (18, 19). The disease usually presents as aseptic pustular lesions following a chronic course, going through different stages in the form of yellow scales or crusts, and at a later stage brown macular residual lesions. The onset of PPP usually occurs in adulthood, severely impairs patients’ quality of life in severe cases, and may associate with extracutaneous involvement, such as nail disease, arthritis, and, rarely, with neutrophilic cholangitis (10). Occasionally PPPP may associate with autoimmune conditions, such as thyroiditis, although it is not known if the prevalence of clinical autoimmunity is higher than in the general population (20). One particular axial spondyloarthritis feature observed in patients with PPPP led to the definition of a syndrome called SAPHO (synovitis, acne, pustulosis, hyperosteosis osteomyelitis) (16). This is characterized by painful swelling of sternocostal and manubrial areas, and its diagnosis is usually established by bone scintigraphy (21).
While GPP typically follows a relapsing, intermittent course with disease-free intervals that may last for months or sometimes years, the evolutive pattern of PPPP is usually chronic and, like other pustular psoriatic subtypes, may associate with PV. It may also combine in some patients with ACH or, rarely, with GPP (13, 16).
Fig. 2. Typical lesions of palmoplantar pustular psoriasis in 3 different patients free of any mutation in IL36RN, CARD14 and AP1S3 genes. (A) Pustular lesions involving palmar areas of hands, with some degree of acropustular damage, reflecting the possible association between palmoplantar pustular psoriasis and acrodermatitis continua of Hallopeau (ACH). (B) Disseminated pustules of the soles leave dark-brown macular lesions, coexisting with fresh evolutive pustules and erythemato-squamous lesions. (C) Typical lesions of ACH involving the toes, leading to destruction of the nail apparatus.
The ACH subphenotype is defined by pustular lesions involving extremities of the hands and feet, with progressive destruction of the nail apparatus, with or without underlying bone erosions (Fig. 2C) (22). Like PPPP, this rare, debilitating form has been reported in between GPP flares in patients with deficiency of IL-36 receptor antagonist (DITRA) (13). The threat of definitive nail and/or bone damage warrants early treatment of patients with ACH.
Diagnosis of pustular psoriasis relies mainly on clinical features and is usually easy. Characteristic histopathological findings include the formation of intraepidermal neutrophilic abscesses, with marked dermal infiltrate composed of neutrophils, monocytes and T-lymphocytes (1, 2). One major differential diagnosis of GPP is acute exanthematous generalized pustular eruption (AGEP), the clinical signs and symptoms of which may be impossible to differentiate from GPP, but which is caused by drugs, notably by anti-infectious chemotherapy, such as pristinamycin and amoxicillin, but also other classes, such as non-steroidal anti-inflammatory drugs, among others (23, 24). The recent detection in patients with AGEP of mutations in IL36RN, sometimes identical to the ones identified in patients with GPP/DITRA, challenges the current view of AGEP and GPP being separate entities (25).
The extreme severity of these inflammatory pustular skin disorders, especially GPP, and the existence of Mendelian familial cases, raised the hypothesis of a monogenic model, unlike most cases of PV. This monogenic model has been robustly established by the identification of homozygous or composite heterozygous, loss-of-function mutations of the IL36RN gene, which encodes a negative regulator of the IL-36 pathway, which is involved in the limitation of the intensity of skin and systemic innate immune responses. Indeed, IL36RN mutations have been found in sporadic or familial cases of GPP in patients from different geographical territories worldwide (13, 26–32). These IL36RN mutations are more prevalent in patients with GPP without plaque psoriasis, and influence the age of disease onset (32). Mutations of IL36RN lead to major structural and functional impairments of its encoded protein, the IL36 receptor antagonist (IL36Ra), leading to increased inflammatory responses resulting from unrepressed interactions of the IL36 pathway agonists IL36α, IL36β and IL36γ with their receptor, and from subsequent uncontrolled activation of the transcription factor NFκB (13). This results in the massive release by keratinocytes, macrophages and dendritic cells, of several inflammatory mediators including CXCL8, TNFα, IL1 and IL23 (33). Dysregulated activation of the IL-36 pathway has also been shown to trigger the expansion and activation of TH17 cells in GPP (34). So far, different scale studies of cohorts from various geographical territories have reported various prevalences of IL36RN mutations, ranging from approximately 5% to 70%, while much lower prevalences have been observed in patients with PPPP, and no causal IL36RN mutation has been detected in patients with PV without pustular psoriasis (29–32, 34). An interesting finding has been the identification of identical IL36RN mutations across the different subtypes of pustular psoriasis (35). However, mutations leading to the absence of IL-36Ra protein expression are preferentially associated with the most severe entities of GPP and AGEP, while hypomorphous mutations seem to be more prevalent in PPPP and ACH (35). The major breakthrough in the identification of causal mutations of the IL36RN gene has been instrumental in establishing without ambiguity the autoinflammatory nature of GPP, and led to the definition of a new entity called DITRA, which differs from the previously described deficiency of IL-1 receptor antagonist (DIRA) by the presence of striking lesions of joints and bones (13, 37, 38). Finally, although causal mutations of IL36RN have not been found in patients with PV, several studies have shown deregulation of the IL-36 pathway in PV lesions (39).
The 2 other genes associated with pustular psoriasis so far are CARD14 and AP1S3. Likewise, heterozygous gain-of-function mutations of CARD14 (caspase activating recruitment domain, member 14), a gene expressed in keratinocytes the protein of which interacts with Bcl 10, a positive regulator of NFκB activation, has been shown to be primarily involved in autosomal dominant forms of PV and in some patients with pityriasis rubra pilaris (40–43). The Adaptor Related Protein Complex 1 subunit sigma 3 (AP1S3) gene has been also found to be heterozygously mutated in patients with different subtypes of pustular psoriasis, mainly GPP and ACH, leading to structural and functional alterations of the protein, a member of the Adaptor Protein 1 (AP1) family, contributing to deregulation of skin innate immune responses (42, 44, 45). Likewise, it is notable that some patients have “digenic” features, e.g. a pattern characterized by mutations reported to be damaging in 2 of the 3 genes identified so far (32). Further identification of other genes, especially in GPP, will undoubtedly complement the current genetic map of pustular psoriasis, and is likely to greatly contribute to personalized therapeutic approaches.
The low prevalence of pustular psoriasis and the capricious course of the disease with unpredictable flaring frequency in many cases of GPP, explain the low level of scientific evidence regarding treatment efficacy. Indeed, although topical steroids and/or vitamin D derivatives, used as single agents or combined, or phototherapies are still used in mild forms of pustular skin disease with limited involved body surface area, pustular psoriasis often requires systemic therapy. In PPPP, cyclosporine has the highest level of evidence for efficacy, while there is weak or very weak evidence, respectively, for acitretin and methotrexate (46–48). More recently, randomized, placebo-controlled phase 3 clinical trials have been conducted in PPPP with secukinumab and guselkumab, targeted inhibitors of IL17A and IL23p19, respectively (49, 50). However, neither drug showed clinically relevant superiority over placebo at the population level, suggesting that the IL23/IL17 pathway is not the major pathogenic axis in pustular disease (49, 50). Randomized clinical trials are currently being conducted in PPPP with inhibitors of cytokines of the IL-1 family, the most advanced programme investigating the efficacy and safety of anakinra, the recombinant form of the IL-1 receptor antagonist, based on encouraging responses in isolated cases, including with ACH (51, 52).
There is even less available evidence in GPP, due to the previously exposed challenges, but also to the spontaneously self-remitting evolutive pattern of acute GPP flare. Thus positive responses reported with conventional or biological drug interventions in the setting of retrospective, or open-labelled prospective trials, should be considered with caution. Therefore, although high-dose steroids, cyclosporine, acitretin and apheresis have been promoted for severe acute flares, and although some biologics, such as IL17 inhibitors, have been approved for GPP in Japan, these interventions lack randomized controlled studies to assess the magnitude of their efficacy effect (53, 54). Furthermore, the efficacy of anakinra, the recombinant form of the IL-1 receptor antagonist, has been reported only in a case series of GPP with or without DITRA, reporting most often transient and partial responses (55, 56). These cases should be confronted with the outstanding efficacy of IL-1 inhibitors in patients with DIRA, emphasizing the fine specificity of pathogenic pathways across different monogenic autoinflammatory syndromes of the skin (36). Therefore, the emerging development of specific inhibitors of the IL-36 pathway in GPP and PPPP is not surprising. The most advanced development investigates an anti-IL-36 receptor monoclonal antibody, which, administered as a single intravenous dose, proof-of-concept study in acute GPP, showed very encouraging results in 7 patients, only 3 of whom were carrying IL36RN mutations (57). Ongoing phase 2 and 3 studies will provide a more accurate picture of the efficacy and safety of this new targeted strategy.
Pustular psoriasis is a very challenging spectrum of autoinflammatory skin diseases, with both clinical and genetic heterogeneity. However, the increasing collaboration between medical experts and scientists is encouraging in enabling the better nosological classification of subentities, as well as the development of specific therapeutic strategies, approximately 100 years after the pioneering description of GPP by von Zumbusch (58).
HB had paid activities as advisor, speaker or consultant for Abbvie, Almirall, Amgen, Biocad, Boehringer-Ingelheim, Celgene, Eli-Lilly, Janssen, Leo Pharma, Mylan, Novartis, Pfizer, Sun Pharmaceuticals and UCB.


 Click to show fullsize
Click to show fullsize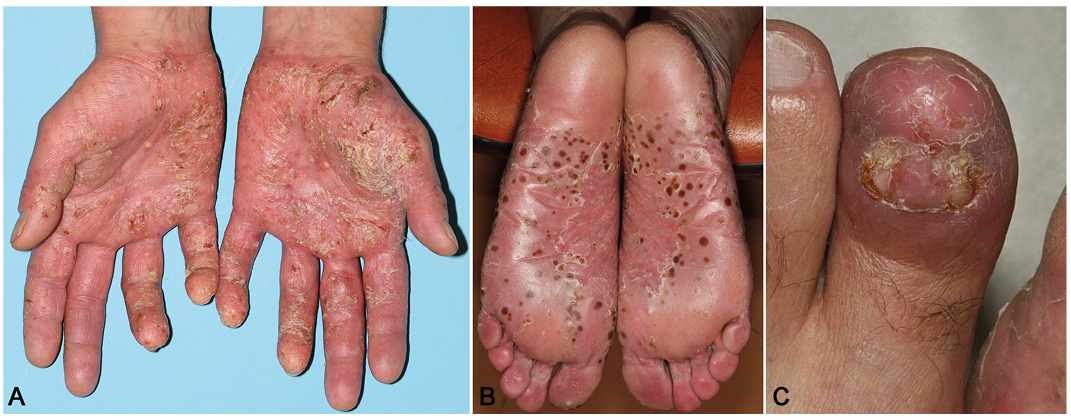 Click to show fullsize
Click to show fullsize