


Department of Dermatology, Hokkaido University Graduate School of Medicine, Sapporo, Japan
Collagen XVII (COL17) is a hemidesmosomal transmembrane protein in the skin, which, in several auto-immune blistering skin diseases, may be targeted by autoantibodies. In addition, loss-of-function mutations in the COL17A1 gene induce a subtype of junctional epidermolysis bullosa. The extracellular domain of COL17 can be physiologically cleaved from the cell surface by ADAM family proteins in a process known as ectodomain shedding. COL17 ectodomain shedding is thought to be associated with the migration and proliferation of keratinocytes. Furthermore, the C-terminal cleavage of COL17 may be associated with basement membrane formation. COL17 can be targeted by various proteases, including MMP9, neutrophil elastase, plasmin and granzyme B, which may be associated with blister formation in pemphigoid diseases. Interestingly, cleavage of COL17 may induce neoepitopes on the proteolysed fragments, and such induction is associated with dynamic structural changes. This review summarizes the current understanding of cleavage of COL17, and how such cleavage relates to blistering skin diseases.
Key words: ectodomain shedding; BP180; bullous pemphigoid; linear IgA bullous disease; epidermolysis bullosa.
Accepted Dec 18, 2019; Epub ahead of print Feb 6, 2020
Acta Derm Venereol 2020; 100: adv00054.
Corr: Wataru Nishie, Department of Dermatology, Hokkaido University Graduate School of Medicine, N15W7, Kita-Ku, Sapporo 060-8638, Japan. E-mail: nishie@med.hokudai.ac.jp
Collagen XVII (COL17, also known as BP180) is an important molecule, which maintains stable adhesion between the dermis and epidermis. Genetic and acquired dysfunc-tions of COL17 lead to blistering skin diseases. However, the expression of COL17 is tightly regulated, depending on various settings, including wound-healing, proliferation and differentiation. Dysregulation of COL17 processing may be associated with the development of blistering skin diseases; thus, it is important to understand the mechanism by which COL17 is processed and the diseases associated with such processing.
Type XVII collagen (COL17), also known as BP180/BPAG2, is a type-II-oriented transmembrane collagen composed of 3 identical 180-kDa α-chains (1). COL17 is one of the hemidesmosomal components of basal keratinocytes. It links keratin intermediate filaments to the underlying dermis via plectin, BP230, laminin 332 and type VII collagen (2). Loss-of-function mutations in the COL17A1 gene result in a subtype of junctional epidermolysis bullosa (JEB), which clinically manifests as blister formation and abnormalities of the hair and teeth (3). Since JEB associated with COL17A1 gene mutations shows a relatively mild phenotype, the disease was previously called “generalized atrophic benign epidermolysis bullosa (GABEB)”.
Autoimmunity to COL17 induces bullous pemphigoid (BP), a major autoimmune blistering skin disease, which commonly develops in elderly people (4, 5). In BP, itchy urticarial erythema and tense blisters develop on the entire body, and the mucous membranes may be involved. Major epitopes for BP autoantibodies cluster tightly within the juxtamembranous extracellular non-collagenous 16th A (NC16A) domain of COL17 (6), and previous studies have revealed the pathogenicity of immunoglobulin G (IgG)-class autoantibodies directing this region (7, 8). COL17 may also be targeted by autoantibodies in other autoimmune blistering skin diseases, including mucous membrane pemphigoid (MMP) and linear IgA bullous disorder (LABD) (4).
The two COL17-associated blistering disorders, JEB (GABEB) and BP, suggest that COL17 is a functionally important structural molecule that maintains stable adhesion between the dermis and the epidermis at the dermal-epidermal junction (DEJ). However, basal keratinocytes are dynamic, and they migrate or differentiate in a context-dependent manner. Therefore, processing of COL17 may be involved in various physiological settings. In addition, dysregulated processing of COL17 may be associated with blistering skin diseases. This review summarizes the current understanding of COL17 processing and the blistering skin diseases associated with such processing.
COL17 ectodomain is constitutively cleaved within the NC16A domain
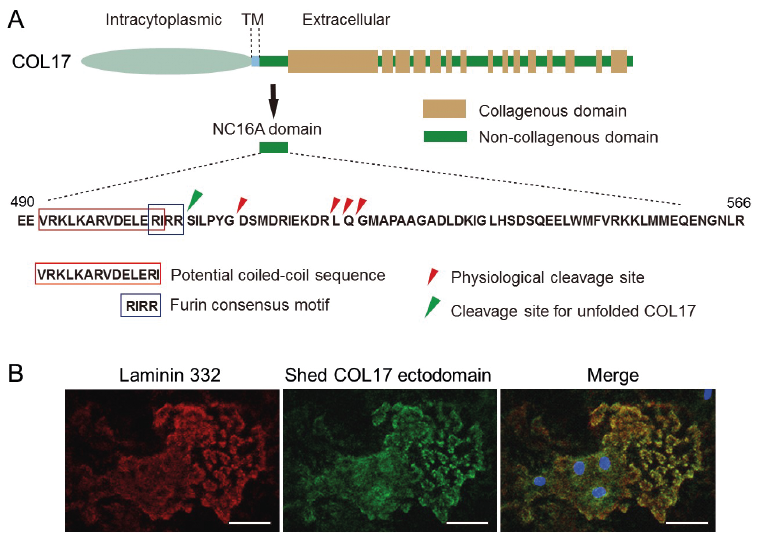
In cultured keratinocytes, the 120-kDa extracellular domain of COL17 is constitutively shed from the cell surface and is detectable in soluble form in culture medium (9, 10). COL17 ectodomain shedding is mediated by ADAM 9, 10 and 17 (11), and mass spectrometry analyses have revealed that the cleavage occurs at different regions within the NC16A domain (Fig. 1A) (12, 13). The results are consistent with the nature of ADAM family proteins, which cleave substrate proteins more preferentially, based on the distance from the cell surface rather than on amino acid sequences. The detection of cleavage sites within the NC16A domain of COL17 enables the production of cleavage-site-specific antibodies specifically detecting the cleaved COL17 ectodomains. Unique antibodies have revealed that migrating normal human keratinocytes cleave COL17 ectodomains, which co-localize with laminin 332 (Fig. 1B), (14) and cleaved ectodomain fragments exist in the DEJ of normal human skin (13, 15). Interestingly, the cleavage site(s) of COL17 in pathological settings may differ from that in physiological settings (15). In genetically manipulated mice whose NC16A domain includes amino acid sequences that impair ectodomain shedding, the inhibition of COL17 ectodomain shedding somewhat accelerated re-epithelialization after skin wounding (16). The suppression of re-epithelialization by COL17 ectodomain shedding is associated with dampening of mTOR signalling (17). However, wound healing processes differ greatly between humans and mice, with wounds in mice healing mainly by contraction (18). Therefore, further studies are essential to address the physiological roles of COL17 ectodomain shedding in human skin.
Fig. 1. Collagen XVII (COL17) processing in physiological settings. (A) Schematics of COL17 and sequences of the NC16A domain. (Copyright 2010: The American Association of Immunologists, Inc.). (B) The shed COL17 ectodomain (green: antibody HK139) and laminin 332 (red: antibody 6F12) co-localize in the extracellular matrix of normal human skin. TM: transmembrane. Blue: DAPI. Scale bar: 40 μm. The figures have been partially modified from previous studies (13, 14). Permission given by publisher.
C-terminal cleavage of COL17
The cleaved 120-kDa ectodomain of COL17, also called as linear IgA dermatosis antigen (LAD-1) , may be further processed at the C-terminal region around the NC4 domain, which migrates around 97 kDa (19, 20). The 97-kDa processed COL17 polypeptide is called linear IgA bullous disorder (LABD)-97 (Fig. 2A). Although it remains uncertain whether LABD-97 is present in normal human skin, C-terminal processing is expected to be physiologically associated with correct basement membrane formation in skin, as described later in this article.
Fig. 2. Neoepitope development in the cleaved collagen XVII (COL17) ectodomains. (A) Schematics of linear IgA dermatosis type 1 (LAD-1) and linear IgA bullous disorder (LABD)-97 polypeptides. (B) LAD IgA-class autoantibodies show more intense reactivity to the cleaved COL17 ectodomains LAD-1 and LABD-97 than to full-length COL17. Note that LAD sera numbers 2, 5 and 10 react more strongly to LABD-97. N: normal control. The immunoblotting data have been partially modified from a previous study (23). BP: bullous pemphigoid. Permission given by publisher.
Cleavage in unfolded COL17
Within the NC16A domain, COL17 has a distinct furin consensus sequence: ‘’RIRR’’. Early studies have suggested that ectodomain shedding of COL17 may be induced by this distinct motif; however, the furin consensus motif is not used under physiological settings (10). What is the physiological role of the furin consensus motif in COL17? As illustrated in Fig. 1A, there are potential coiled-coil sequences just before the furin consensus motif, and these sequences initiate the folding of COL17 as a collagen triple helix in the N to C direction (21). When coiled-coil disruptive mutations are introduced, COL17 folding is impaired and unfolded COL17 accumulates in cells. The unfolded COL17 is cleaved by furin at the ‘’RIRR’’ furin consensus motif in the Golgi apparatus before being incorporating into the cell membrane. Finally, cleaved 120-kDa ectodomains derived from unfolded COL17 are expelled from the cells (21). Thus, cleavage at the furin consensus motif within the NC16A domain of COL17 is important for maintaining the quality of the molecule.
Cleavage within the NC16A domain induces neoepitopes in processed COL17 ectodomains
As described, cleavage of COL17 within the NC16A domain yields a 120-kDa ectodomain polypeptide, known as LAD-1 (Fig. 2A). LAD-1 partially contains sequences of the NC16A domain, with which BP autoantibodies preferentially react (12, 13). Similarly, MMP autoantibodies targeting the C-terminal regions of COL17 may react with LAD-1. It is notable that, in some cases of BP and in many cases of LABD, autoantibodies show more preferential reactivity to LAD-1 than to full-length COL17 (Fig. 2B) (19, 22), indicating that cleavage within the NC16A domain induces neoepitopes on the cleaved LAD-1. In silico predictions based on detected cleavage sites reveal that the antigenicity of the remnant NC16A sequences in the cleaved COL17 ectodomains increase despite the different cleavage sites (13). Furthermore, monoclonal antibodies target the 15th collagenous (COL15) domain with preferential reactivity to LAD-1, suggesting that cleavage within the NC16A domain induces dynamic structural changes in COL17 (23).
C-terminal cleavage of COL17 induces neoepitopes on the LABD-97 fragment
Since LABD autoantibodies react more preferentially with LAD-1 than with full-length COL17, they usually have strong reactivity to LABD-97 (Fig. 2B) (24). Interestingly, LABD autoantibodies may have greater reactivity to LABD-97 than to LAD-1 (Fig. 2B), suggesting that C-terminal cleavage has additional effects on neoepitope development (23). A previous study reported that epitopes on the 15th collagenous domain appear after C-terminal cleavage (23), which is consistent with an epitope mapping study of LABD autoantibodies (25).
COL17 cleaving enzymes in bullous pemphigoid
In BP lesional skin and blister fluid, several proteolytic enzymes are known to exist, including plasmin, neutro-phil elastase and MMP-9 (4, 5). In vitro studies have revealed that neutrophil elastase (26), MMP-9 (27) and plasmin (19) are able to cleave COL17. Of these, plasmin is known to cleave within the NC16A and NC4 domains of COL17 ectodomains, yielding 120-kDa LAD-1 and 97-kDa LABD-97 fragments (19, 20, 28). In addition, a recent study has reported that granzyme B, a serine protease secreted by immune cells, is highly expressed in infiltrated cells in BP lesional skin and that not only does this enzyme cleave COL17, but it also cleaves other molecules present at the DEJ, including α6/β4 integrins and collagen VII (29).
Impaired C-terminal cleavage of COL17 may induce disorganized basement membrane formation
When homozygous R1303Q mutations occur in the COL17A1 gene, a mild and localized form of JEB develops that is clinically characterized by mechanical blisters, tooth and nail abnormalities, and sclerotic fingers associated with a loss of fingerprints (Fig. 3) (30, 31). Histopathologically, duplication of the basement membrane is characteristic of JEB patients with R1303Q mutations (Fig. 3B). Since R1303Q mutations impair the C-terminal processing of COL17, such processing is thought to be essential for normal basement membrane formation in skin (28).
Fig. 3. Collagen XVII (COL17) R1303Q mutation induces blistering disease associated with disorganized basement membrane formation. (A) The R1303Q mutation is located within the NC4 domain. (B) A previously reported 32-year-old COL17 R1303Q+/+ patient (28). The arrow indicates a mechanical blister. (C) A disorganized and duplicated basement membrane is a characteristic histopathological feature, which can be detected by anti-type IV collagen antibodies (PHM-12+CIV22). (D) Western blotting using anti-COL17 NC16A antibodies (NC16A-3) on extracellular matrix proteins derived from mal human epidermal keratinocytes (NHEKs) and keratinocytes from a R1303Q+/+ junctional epidermolysis bullosa patient. The arrow indicates that linear IgA bullous disorder (LABD)-97 is absent in R1303Q+/+ keratinocytes, suggesting that the C-terminal cleavage of COL17 is impaired. The figures have been partially modified from previous studies (28). LAD-1: linear IgA dermatosis type 1. Permission given by publisher.
Impaired cleavage of COL17 may induce the breaking of tolerance to bullous pemphigoid autoantigens
BP is induced by autoantibodies targeting the hemidesmosomal components COL17 and/or BP230. Although the pathomechanism of autoantibody-dependent blister formation has been studied extensively, there has been no full elucidation of why tolerance to these autoantigens may be broken in certain individuals. Immune tolerance to molecules may be broken by various triggering events, including thermal burns, ultraviolet (UV) irradiation and surgery (32). In addition, recent studies have reported that anti-type II diabetes mellitus drug dipeptidyl peptidase IV inhibitors (DPP4i) are a risk factor for the onset of BP (33, 34). Furthermore, impaired Treg function may break the tolerance to COL17 and BP230 (35, 36). However, it remains unclear whether the impaired expression of pemphigoid autoantigens may induce the breaking of tolerance. In 2015, Hurskainen et al. (37) produced a genetically manipulated mouse lacking the immunodominant NC14A domain of Col17, a domain that corresponds to the human NC16A domain of COL17. Since NC14A is essential for the ectodomain shedding of mouse Col17, this is another shedding-deficient model. It is notable that the mice are prone to scratching themselves and spontaneously developed anti-Col17 autoantibodies, although no blistering was observed. Whether impairments of BP autoantigens induce the breaking of tolerance had not been elucidated, therefore this study brought important information on the pathomechanism behind the breaking of tolerance to COL17.
Cleaved fragments on immune cells in bullous pemphigoid lesional skin
The roles of IgG-class anti-COL17 autoantibodies in the development of blisters have been studied extensively; in contrast, the pathomechanism for urticarial erythema has not been fully elucidated. Previous studies have reported that both IgG- and IgE-class anti-COL17 NC16A autoantibodies are present in BP sera (38, 39). Although in vivo IgE deposition at the DEJ may be observed in BP, the positivity rate is not high (40). Notably, Freire et al. recently reported that IgE is rarely observed at the DEJ, but that it is prominent on mast cells and eosinophils in the dermis, in which COL17 ectodomain fragments co-localized with IgE (39). This observation is consistent with the fact that the shed COL17 ectodomain is soluble after being cleaved from the cell surface, as described previously.
JEB and pemphigoid diseases have proven that COL17 is a vital player in the stable adhesion between the dermis and epidermis at the DEJ in the skin. However, this adhesion needs to be tightly regulated in a context-dependent manner, for basal keratinocytes to migrate, differentiate and proliferate. Undoubtedly, the processing of COL17 is involved in various normal physiological, as well as pathological, settings, and will be the focus of future study.
The author has no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize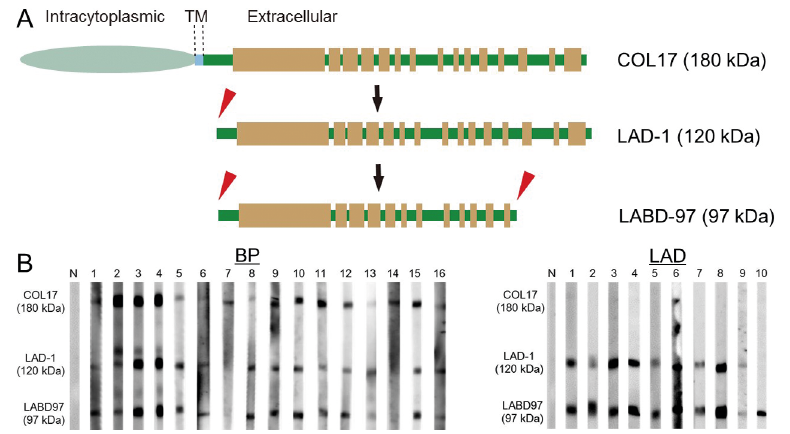 Click to show fullsize
Click to show fullsize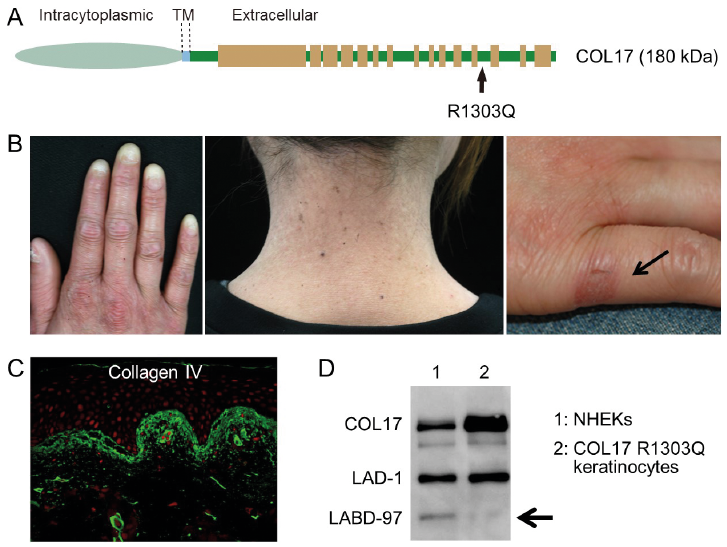 Click to show fullsize
Click to show fullsize