


Lübeck Institute of Experimental Dermatology and Center for Research on Inflammation of the Skin, University of Lübeck, Lübeck, Germany
Pemphigoid diseases are organ-specific autoimmune diseases of the skin and/or mucous membranes. They are caused by autoantibodies targeting adhesion molecules located at the dermal–epidermal junction. While the diagnostics of pemphigoid diseases and insights into their pathogenesis have improved significantly, the development of novel treatments that are effective and safe remains an unmet medical need. However, numerous pre-clinical studies and early clinical trials have recently been launched. This review summarizes some pathways leading to drug development in pemphigoid diseases, namely: (i) hypothesis-driven drug development; (ii) omics-based drug development; (iii) drug repurposing; (iv) screening-based drug development; and (v) drug development based on careful clinical observations. Ultimately, it is hoped that this will lead to personalized and curative treatments.
Key words: bullous pemphigoid; epidermolysis bullosa acquisita; translational medical research; disease models; animal autoantibodies.
Accepted Dec 18, 2019; Epub ahead of print Feb 6, 2020
Acta Derm Venereol 2020; 100: adv00055.
Corr: Ralf J. Ludwig, Lübeck Institute of Experimental Dermatology University of Lübeck, Ratzeburger Allee 160, DE-23538 Lübeck, Germany. E-mail: ralf.ludwig@uksh.de
Despite detailed insights into the pathogenesis of pemphigoid diseases, their treatment still relies on unspecific immunosuppression. Since such treatment contributes significantly to the high patient morbidity and increased mortality, we propose pathways that may facilitate drug development for pemphigoid diseases. With this we aim to foster translational research to develop new treatment strategies for patients with pemphigoid diseases.
(Muco)-cutaneous blistering is the clinical hallmark of pemphigoid diseases (PD). They are characterized and caused by autoantibodies targeting adhesion molecules located at the dermal–epidermal junction. Depending on clinical presentation, the specificity and isotype of the autoantibodies in the following PD can be distinguished (1):
Treatment of all PD centres on unspecific, systemic immunosuppression, whereby corticosteroids are usually the first line of treatment. Among PD, PG, LAD and p200 usually respond well to treatment and long-term remissions are common. Likewise, BP also responds well to either systemic or topical corticosteroids. However, after withdrawal of treatment, BP relapses in almost 50% of patients within 6 months, requiring long-term corticosteroid treatment, which contributes to patient morbidity and mortality. Both, MMP and EBA are notoriously difficult to treat, and often remission is achieved only after months of immunosuppressive therapy, usually a combination of several drugs (1, 11–16).
This “need for better treatment options” has been identified recently by patients and physicians in a survey to identify the medical need in PD (17). In addition to the current limitations regarding treatment options, the increasing incidence of PD, especially in ageing societies (18, 19), further contributes to the medical need to develop novel treatment strategies for PD that are both effective and safe. This increasing medical need has also prompted a significant number of translational studies and clinical trials in PD (20, 21). Unfortunately, however, these clinical trials will not fully address the medical need in PD. Thus, ongoing translational research is required to continuously improve the treatment options, ultimately aiming for personalized and curative treatment.
There are many pathways that may contribute to drug development in PD (Table I, Fig. 1). While it may be simplistic, it could be useful to categorize these pathways to new drugs, as follows: (i) hypothesis-driven drug development; (ii) omics-based drug development; (iii) drug repurposing; (iv) screening-based drug development; and (v) drug development based on careful clinical observations. Examples of each of these pathways to novel treatments for PD are given and discussed in more detail below. The aim of this review is to promote drug development for patients with PD by providing these examples. Another important aim of this article is to initiate a discussion on how this goal is best achieved. Hence, the authors are looking forward to comments from the community, which it is hoped will lead to a fruitful discussion.
Table I. Examples of drugs evolving from the outlined pathways to drug development in pemphigoid diseases
Fig. 1. Pathways to new drugs for the treatment of pemphigoid diseases. (a) Development of new pemphigoid treatments based on hypothesis-driven research. As an example, the development of new complement inhibitors, such as anti-C1s antibodies and coversin are depicted. Based on the clinical observation that complement deposits (in green) are highly prevalent in the skin of patients with pemphigoid disease (PD) (1) and the observation that mice deficient (ko) in specific complement proteins are protected from the induction of experimental PD (34), targeting complement activation was assumed to have disease-modifying effects in PD. Both (in vitro assays, middle panel) and a phase I clinical trial demonstrated that anti-C1s impairs/reduced complement deposition along the dermal-epidermal junction. (b) Development of new pemphigoid treatments based on complex data sets and omics. Here biological specimen, i.e. affected vs. non-affected skin from patients or pre-clinical model systems (left-hand image), are subjected to unbiased measurement, for example RNA-sequencing or proteomics. In the example provided, RNA expression in the skin was contrasted between healthy mice and mice with EBA. Subsequently (middle image), data analysis is performed, leading to the identification of potential pharmacological targets, such as Sykb. For functional validation, in vitro systems or pre-clinical model systems (right-hand image) may be used. (Example from Samavedam et al. (37)). (c) Development of new pemphigoid treatments based on drug repurposing. Already licensed drugs, for indications than other pemphigoid, can be repurposed for PD. The rationale for drug repurposing in pemphigoid can either be based on clinical observations, i.e. case report series that a given drug is also effective in pemphigoid, such as doxycycline, or be hypothesis-driven, as shown for dimethyl fumarate, which has a long history as an anti-psoriatic agent (left-hand panel), which also ameliorates experimental PD (right-hand panel). (d) Development of new pemphigoid treatments based on drug screening. If putative defined drug targets are not known, drug screening can be performed in in vitro model systems, which are up-scalable and highly reproducible. In PD, examples for these assay systems are immune complex-induced release of reactive oxygen species from neutrophils, anti-CD3/CD28-induced T cell proliferation, IL-21/antiCD40L-induced B cell proliferation and anti-BP180 IgG-induced cytokine release from keratinocytes (51). Drug libraries, for example the Prestwick Chemical Library (66) or the FDA-approved Drug Library from Selleckchem, can be obtained commercially. After the initial screening the identified potential drugs need to be validated in vitro and in vivo (left-hand panel). (e) Development of new pemphigoid treatments based on clinical observations. After the identification of the pathogenic relevance of autoantibodies in PD and the clinical observation of a correlation of the levels of the circulating autoantibody titres with disease severity, immunoadsorption/plasmapheresis were introduced to the management of PD. However, immunoadsorption is limited because all antibodies are removed. Hence, the procedure has to be paused, and does not elute all autoantibodies from the patients. Using the insights from detection of specific autoantibodies in PD, first attempts were made to develop antigen-specific immunoadsorption.
Hypothesis-driven drug development: anti-C1s antibodies in bullous pemphigoid
Complement deposition at the dermal–epidermal junc-tion is one of the diagnostic pillars of PD (22). The functional contribution of complement to the pathogenesis of PD has been well documented in pre-clinical model systems (23, 24). Recent data, however, suggests that complement has a more complex role in pemphigoid, whereby some complement receptors confer protection from development of clinical disease (25), or where PD develops independent of complement activation (26). Nonetheless, the complement component C5a has to be considered as one of the main drivers of autoantibody-induced tissue damage in PD (27, 28).
Based on these considerations, function-blocking antibodies to C1s, which initiate the classical complement activation cascade, were developed (29). These anti-C1s antibodies, dose-dependently inhibited the immune complex-induced complement fixation on human skin cryosections (30). More recently, a phase I clinical trial in patients with BP was successfully completed, in which the anti-C1s antibody TNT009/BIVV009 was found to be safe and tolerable in this elderly population, with only mild to moderate adverse events (31). Furthermore, a phase II clinical trial using the dual C5/LTB4 inhibitor coversin is currently being conducted in BP, with promising initial data (32). What is perhaps most striking about the clinical development of these 2 complement inhibitors is the long time needed to translate the clinical and experimental findings on the importance of the complement system into clinical trials. The presence of complement deposits in BP was discovered in the late 60th of the last century (33), and the central role of the complement system in disease pathogenesis was described over 20 years ago (34).
Interestingly, complement activation in PD seems to be restricted to the skin, where C3 deposits are regularly observed, both in patients and animal models of the diseases. More specifically, plasma concentrations of C3a, C4a and C5a in patients with BP were identical to those observed in age- and sex-matched controls. In the same cohort of patients, concentrations of these complement compounds did not change after clearance of skin lesions. In contrast, all of the patients had C3 deposits in the skin at the time of diagnosis (30). Recently, targeted complement therapeutics have been developed, which preferentially bind to sites where complement is activated (35, 36). These targeted complement therapeutics are expected to be both more effective and have fewer adverse events compared with non-targeted complement inhibitors.
Omics-based drug development: validation of spleen tyrosine kinase as a target for treatment of pemphigoid disease
With the availability of novel technologies; for example, genetics, proteomics and RNA sequencing, an unbiased exploitation of novel therapeutic targets can be performed. Regarding PD, such approaches have, however, been sparsely used, and have been limited to mouse models (7). In detail, contrasting cutaneous RNA expression from mice with and without experimental EBA, several potentially disease-promoting genes were identified, i.e. Sykb, the gene encoding for the spleen tyrosine kinase (SYK). To evaluate the functional role of differential Sykb expression in EBA, experimental EBA was induced in mice that were treated with selective SYK inhibitors, or EBA was induced in SYK-deficient mice. In both experiments, complete protection from induction of experimental EBA was observed if SYK was blocked (37). In parallel, hypothesis-driven research, made similar observations (38). Thus, SYK has been independently identified and validated as a potential therapeutic target for PD.
Unfortunately, however, omics datasets are quite sparse for PD. To the best of our knowledge, only one GWAS has been published so far, reporting an association of MMP with HLA-DQB1*03:01 and rs17203398, in which the intronic region of GALC is located (39). Therefore, in the future, a joint community effort is required to collect well-defined patient samples using standardized procedures for sample acquisition and storage. Alternatively, or in parallel, multi-omics data from model systems (as reported for SYK) may be used for target identification, as well as functional validation. For translation into clinical use, expression of the identified targets may be performed in corresponding patient samples. The advantage of such an approach is that fewer patient samples would be required.
Drug repurposing: doxycycline and dimethyl fumarate for bullous pemphigoid treatment
In dermatology, the use of the anti-CD20 antibody rituximab, initially developed for the treatment of B cell malignancies (40), for the treatment of pemphigus (41) is a good example of drug repurposing. In contrast to “conventional” drug development, already licensed compounds are evaluated for efficacy in other indications. The already known safety profile of the licensed drugs, the decreased time and costs of drug approval are the main advantages of drug repurposing (42).
Regarding PD, the antibiotic doxycycline has recently been demonstrated to be effective in the treatment of BP (43). In a comparative clinical trial, 200 mg of doxycycline, achieved clinical remission in 74% of patients within 6 weeks; while prednisolone (initial dose 0.5 mg/kg) induced remission in 91% of patients. Regarding adverse events, 18% of doxycycline-treated patients experienced a grade 3 or greater adverse event. This was significantly lower, compared with prednisolone, where the number of adverse events was 2-fold higher. Another compound that is currently evaluated for repurposing in BP is dimethyl fumarate (DMF). In Germany, the compound has a long-standing history as an anti-psoriatic agent (44), and more recently has also been licensed for treatment of multiple sclerosis (45). DMF has a multitude of biological effects, including a shift in cytokine expression, a suppression of leukocyte extravasation, anti-oxidant properties, and many others (46). Based on these properties, we hypothesized that DMF may also be beneficial for the treatment of PD. Indeed, treatment of mice with already established clinical EBA manifestations led to a significant improvement in disease activity, while clinical disease severity increased in solvent-treated mice (47). On a molecular level, the beneficial effects of DMF in EBA are mediated through the hydroxycarboxylic acid receptor 2 (48). Based on these findings, the DPem consortium was established to evaluate the safety and efficacy of adjuvant DMF in BP patients responsive to corticosteroid treatment. Centres in France, Poland, Turkey and Germany will recruit 210 patients with BP and allocate these to DMF or placebo.
To the authors’ knowledge there are additional drugs soon to be published that have the potential for repurposing in PD. We expect that this pathway to novel drugs for PD will lead to the approval of several new treatment options for pemphigoid patients, using “old” drugs from other indications.
Drug screening
The use of chemical libraries to identify inhibitors of specific molecules, or the use of complex, but up-scalable, model systems is well established for drug development (49, 50). While the use of specific (enzymatic) assays is very well suited to identify new compounds for known pharmacological targets, the use of complex, up-scalable systems in chemical screens offers advantages in instances where molecular defined targets are not known. Despite the fact that up-scalable complex in vitro models of PD are already established (51), these have, so far, not been used for drug development in pemphigoid. Examples of these up-scalable model systems are immune complex-induced release of reactive oxygen species (ROS) from neutrophils, or autoantibody-induced cytokine release from keratinocytes, as well as stimulation of T cells using anti-CD3/CD28 and B cell stimulation with IL-21 and anti-CD40L (51, 52).
An envisioned work-flow of such an approach would be to screen compounds of a chemical library to inhibit activation of immune cells or autoantibody-induced cytokine release from keratinocytes with a relatively small sample size. Candidate compounds would be selected based on pre-defined cut-off criteria. Subsequently in vitro and in vivo validation (using appropriate animal pre-clinical model systems (53), would be employed before clinical trials.
It is hoped that these models, as well as computational approaches to drug development, such as the Connectivity Map (54), will lead to the identification of novel compounds suited for the treatment of PD.
Clinical observations: immunoadsorption for bullous pemphigoid
The detection of IgG deposits along the dermal–epidermal junction in PD (55) and the identification and cloning of the corresponding autoantigens (56) led to the development of serological test systems for the diagnosis of PD (1). This, by itself, is a good example, of how clinical observations and basic research can improve diagnosis. In addition, insights into the pathogenetic role of these autoantibodies (24) prompted the use of immunoadsorption/plasmapheresis in PD (57). More recently, 2 case series have been published, reporting the outcome of immunoadsorption in 26 patients with BP. Interestingly, and in contrast to other autoimmune skin blistering diseases, such as pemphigus, long-lasting remissions were observed in the majority of patients (58, 59). This data, however, should be interpreted within the limitations of case series, as well as the use of concomitant treatments.
Currently, removal of autoantibodies by immunoadsorption is, however, limited because all antibodies are removed, rather than selective removal of autoantibodies. Hence, vigorous and prolonged removal cannot be performed using unspecific immunoadsorption. In mice, at least, this limitation has been overcome: by using insights on the autoantigens in pemphigus and PD, which are currently exclusively used for diagnosis (22), columns specifically removing autoantibodies targeting the NC16A domain and Dsg3 were developed, and (in part) successfully employed in animal models (60, 61). If these insights from pre-clinical model systems can be translated into clinical use, immunoadsorption will most likely become a more widely used treatment modality for PD. Another, potentially very selective and antigen-based, treatment is the use of chimeric autoantigen receptor (CAAR) T cells, which have been shown to selectively deplete specific autoreactive B cells in mouse models of pemphigus (62).
With the increasing number of clinical trials in PD (21), approval of several new treatments for PD can be expected within the next 3–5 years. However, these trials only recruit patients with BP. For all other PD, to the best of our knowledge, there are currently no ongoing clinical trials, despite the high medical need in MMP and EBA. Therefore, specific, or maybe basket, trials that also include these patients would be highly warranted. Regarding curative treatments, the above-mentioned approaches towards the development of antigen-specific immunoadsorption for BP, or the CAAR-T-cell approach could be tailored to each patients’ autoantibodies. In particular, removing the autoreactive B/plasma cell population could induce long-lasting remission, or even a cure, for PD. While translating these interesting findings from pre-clinical model systems into clinical use will take considerable time, a personalized treatment for PD could be implemented relatively quickly using established diagnostic and therapeutic procedures: in single-centre and retrospective studies, several biomarkers have been identified that indicate relapse in BP; for example, the presence of anti-type VII collagen autoantibodies, variations of the glucocorticoid receptor β, or CXCL10-induced matrix metalloproteinase 9 secretion (63–65). Given, that (some of) these are validated in prospective multicentre diagnostic clinical trials, tapering of immunosuppression could be adjusted to the expression of these biomarkers.
Collectively, the high medical need to develop new treatments for PD has prompted a very exciting new area of translational research in this field, which is expected to improve the treatment of patients with PD in the future. New drug approvals, more clinical trials, and personalized and curative treatments are expected.
This work has been financially supported by the Research Training Group “Modulation of Autoimmunity” (GRK 1727) and the Excellence Clusters “Inflammation at Interfaces” (EXC 306), and “Precision Medicine in Chronic Inflammation” (EXC 2167), all from the Deutsche Forschungsgemeinschaft.
RJL has received research funding from Miltenyi Biotec, Biogen, Biotest, Almirall, True North Therapeutics, UCB Pharma, ArgenX, TxCell, Topadur, Incyte and Admirx and fees for consulting or speaking from ArgenX, Immunogenetics, Novartis and Lilly. KB consults for ArgenX.


 Click to show fullsize
Click to show fullsize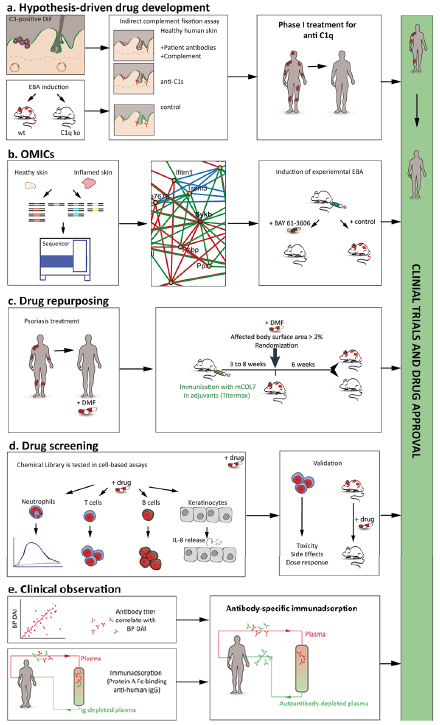 Click to show fullsize
Click to show fullsize