


1Department of Dermatology and Allergology, ERN-Skin, 3Laboratory of Genetics and 7Department of Clinical Genetics and Department of Medical and Clinical Genetics, University of Helsinki and Helsinki University Central Hospital, 2Institute for Molecular Medicine Finland (FIMM), Helsinki Institute of Life Science (HiLIFE), University of Helsinki, 4Folkhälsan Research Center, Helsinki, Finland and Research Programs Unit, Stem Cells and Metabolism Research Program, University of Helsinki, Helsinki, Finland, 5Department of Biosciences and Nutrition, Karolinska Institutet, Huddinge, 6Science for Life Laboratory, Department of Gene Technology, KTH-Royal Institute of Technology, Solna, Sweden, 8Norwegian Centre for Molecular Medicine (NCMM), University of Oslo, Oslo, Norway, 9Department of Genetics, Université Paris Descartes - Sorbonne Paris Cité, and 10Department of Dermatology and Reference Center for Genodermatoses and Rare Skin Diseases (MAGEC), FIMARAD, ERN-Skin, Université de Paris APHP5, INSERM U1163, Institut Imagine, Hôpital Universitaire Necker-Enfants Malades, Paris, France. E-mail: liisa.harjama@hus.fi
*An abstract of this manuscript was published as a poster at the 19th Annual Meeting of the European Society of Pediatric Dermatology, May, 2019.
Accepted Jan 9, 2020; Epub ahead of print Jan 16, 2020
Acta Derm Venereol 2020; 100: adv00060
Mutations in the Secreted LY6/urokinase-type plasminogen activator receptor (uPAR)-related protein 1 gene (SLURP1) cause the recessively inherited palmoplantar keratodermas (PPK) Mal de Meleda (MDM, MIM 248300) and Gamborg-Nielsen (GN, MIM 244850 (1–5). MDM is characterized by diffuse progressive and transgredient erythematous PPK, typically starting in infancy. Hyperkeratotic plaques on the elbows and knees, nail dystrophy, perioral erythema, brachydactyly, and conical-shaped fingers are reported, as well as, rarely, constriction rings with spontaneous auto-amputation of digits. Hyperhidrosis and bacterial or fungal superinfection can result in malodorous macerations, and reduced mobility of the hands and feet is common (6, 7).
GN is milder than MDM, characterized by a diffuse and transgredient hyperkeratosis with an erythematous border, and sometimes with tapered fingers (1–3, 5). The nails are normal. Distal hyperkeratosis is present only on the knuckle pads (5).
Most MDM patients are homozygous for c.82del p.(Cys28Alafs*5), c.43T>C p.(Trp15Arg) and c.286C>T p.(Arg96*) (4, 7–9), while GN patients are typically homozygous for a missense mutation c.43T>C p.(Trp15Arg) (5). The c.82del p.(Cys28Alafs*5) variant was reported in Croatian, Tunisian and Algerian families and in a Scottish patient, suggesting a founder effect (9). This paper extends the SLURP1 spectrum by describing phenotypic variation in 3 unrelated patients with diffuse PPK, and reporting 2 new SLURP1 mutations.
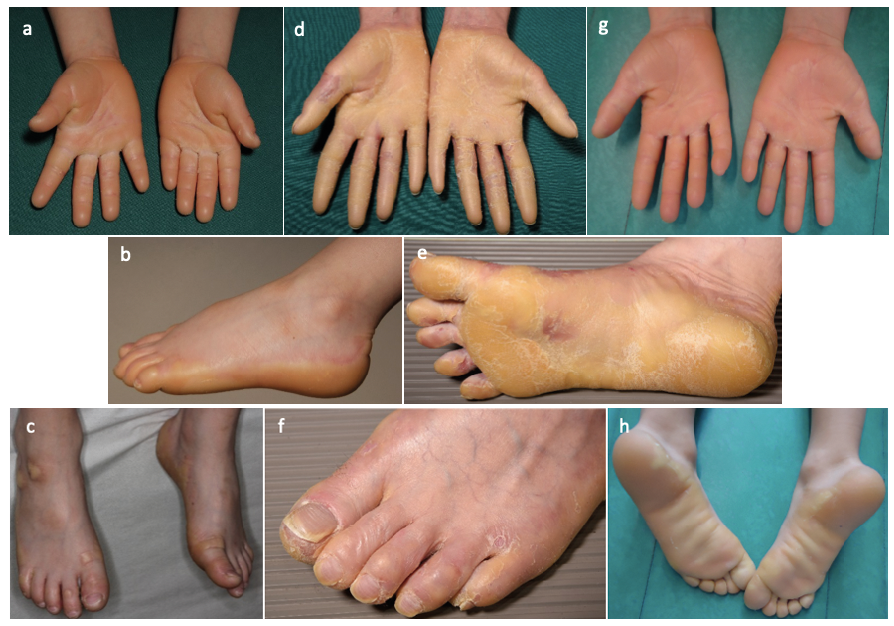
Case 1. An 8-year-old Finnish boy presented with diffuse waxy yellow PPK with a slight erythematous border since infancy (Table SI). Progressive hyperkeratosis was noted on the dorsal surface of his fingers and toes, and on his knees and elbows (Fig. 1 and Fig. S1). Minor wounds left hyperkeratotic scars, and water exposure turned the hyperkeratosis white and spongy. All nails were brittle with longitudinal ridges, and his teeth had enamel defects. Whole exome sequencing (Appendix S1) revealed a maternally inherited missense mutation, c.178G>A p.(Glu60Lys) (rs200727790) in exon 2 and a paternally inherited in-frame deletion c.218_220del p.(Cys73del) (rs748879163) in exon 3 of SLURP1 (Fig. S2).
Fig. 1. Patient phenotypes. (a–c) phenotype of case 1 c.178G>A p.(Glu60Lys) / c.218_220 p.(Cys73del), (d–f) phenotype of case 2 c.178G>A p.(Glu60Lys), and (g, h) phenotype of case 3 c.82del p.(Cys28Alafs*5) / c.286C>T p.(Arg96*).
The p.(Glu60Lys) variant was predicted probably damaging by Polyphen and tolerated by SIFT (10). A total of 29 heterozygous p.(Glu60Lys) carriers, yielding a worldwide population allele frequency of 0.0001037, were reported in the GnomAD population allele frequency database (https://gnomad.broadinstitute. org; last accessed 20 August 2019). There were 20 Finnish carriers (allele frequency 0.0008481). No homozygotes were reported. Only 1 Finnish heterozygous carrier for p.(Cys73del) was reported (GnomAD Finnish population allele frequency 0.000005250, worldwide population allele frequency 0.000004117).
Case 2. A 44-year-old Finnish woman had diffuse, non-progressive PPK since infancy with a waxy yellow hyperkeratosis with an erythematous border (Table SI). The hyperkeratosis expanded on the dorsal surface of her hands and feet, and a hyperkeratotic plaque was also present on the elbow (Fig. 1 and Fig. S1). She had hyperhidrosis of the hands, feet and armpits and frequent dermatophyte infections on the affected skin. All nails were clubbed and hyperkeratotic. On water exposure the affected skin turned white and spongy. A PPK gene panel (Appendix S1) revealed homozygosity for SLURP1 c.178G>A p.(Glu60Lys) (Fig. S2). Haplotype analysis revealed that case 1, his mother, and case 2 shared a haplotype of approximately 3,000 kb (chr8:142544410-145535881 (hg37)) surrounding the c.178G>A p.(Glu60Lys) variant, suggesting common ancestry.
Case 3. An 11-year-old Romanian boy presented with a waxy diffuse PPK with a slight erythematous border without transgrediens and progrediens since the age of 2 years (Fig. 1, Table SI). There was no nail, hair or tooth involvement. A custom gene panel revealed compound heterozygous mutations for a paternally inherited SLURP1 frameshift deletion in exon 2, c.82del p.(Cys28Alafs*5) (rs587776601) and a maternally inherited truncating mutation in exon 3 c.286C>T p.(Arg96*) (rs121908317). The former causes a premature stop codon at amino acid position 32 and the latter results in the deletion of cysteine involved in the highly conserved disulphide bridges (4). Both have previously been reported homozygous in Algerian and Croatian MDM patients (4). Skin biopsies of all 3 cases revealed prominent hyperkeratosis with a very compact cornified layer, epidermal acanthosis with a pronounced granular layer and elongation of the rete ridges (Fig. S3). No conclusive immunohistochemistry was possible for SLURP1.
We report 3 PPK patients with SLURP1 mutations and a phenotype sharing aspects of MDM and GN. Their PPK is characterized by a waxy moderately thick diffuse hyperkeratosis with an erythematous border similar to GN, although cases 1 and 2 have elbow and knee hyperkeratosis with nail abnormalities, which are rarely seen in GN, but rather in MDM. Otherwise, the manifestations are distinct-ly less severe than MDM, lacking, e.g. erythematous inflammation and hyperkeratosis extending widely beyond palms and soles, pseudoainhum, reduction of motility and perioral erythema. Case 3 has the mildest phenotype, with only palmoplantar hyperkeratosis. A novel feature of cases 1 and 2 is the aquagenic whitening of PPK not previously reported with MDM or GN. Our cases share similarities with milder diffuse PPK phenotypes described previously with SLURP1 mutations c.43T>C p.(Trp15Arg) and c.256G>A p.(Gly86Arg) (9, 11, 12). We identified 2 new SLURP1 PPK-associated variants c.178G>A p.(Glu60Lys) and c.218_220del p.(Cys73del), which are located in the central loop II region of SLURP1 (10). In SLURP2 the corresponding loop is critical in a complex formation with its receptors (13). The c.178G>A p.(Glu60Lys) is in the vicinity of the Tyr61–Pro62 peptide bond responsible for the conformational exchange in SLURP1 structure (10). The deleted Cys73 forms a disulphide bond with Cys43 in loop I of SLURP1, which may stabilize the antiparallel β-sheet structures in SLURP1 (10, 14). An 8-fold higher allele frequency of the c.178G>A variant in the Finns indicates an enrichment in Finland and the shared ~3,000 kb haplotype in its carriers suggest a Finnish founder effect.
In conclusion, we further expand the spectrum of SLURP1 mutations with 2 novel mutations c.218_220del p.(Cys73del) and c.178G>A p.(Glu60Lys), the latter being a plausible Finnish founder mutation. Most importantly, the phenotypes of the cases reported here share features of both MDM and GN, thus verifying that these 2 can be seen as a single entity with large phenotypic variability. This suggests that SLURP1 might be involved in PPK mimicking KRT1 or KRT9 mutations.
The authors acknowledge support from the Finnish Dermatological Society’s (SILY) grant and Helsinki University Hospital Research Funds.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize