


1University Hospital Heidelberg, Department of Dermatology, Heidelberg, and 2Dermatopathologie Friedrichshafen, Friedrichshafen, Germany. E-mail: fouad.mitri@med.uni-heidelberg.de
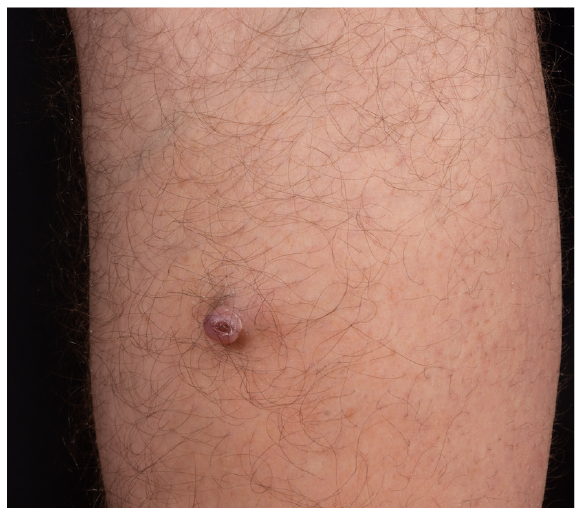
A 69-year-old Caucasian man, under immunosuppressive therapy since 2004 with mycophenolic acid (360 mg twice daily) and sirolimus 2 mg once daily, after a kidney transplantation, presented to our outpatient department with a painless, solitary erythematous nodule on his right calf, measuring approximately 1.2 cm in diameter, with an ulcerated centre (Fig. 1). The nodule had initially appeared 2 years previously and had increased in size until presentation. At first, the patient noted occasional bleeding, which later receded spontaneously. The patient’s medical history included gout and piebaldism. His chronic kidney disease was initially caused by Cacchi-Ricci disease.
The lesion was surgically excised. Macroscopic findings revealed a reddish, partly greyish, gelatinous mass, measuring 3×2.5×1.5 cm. Intraoperatively, myxoid consistency was noted, as well as a relatively deep dermal invasion.
What is your diagnosis? See next page for answer.
Fig. 1. Clinical findings. Firm erythematous nodule with a central ulceration, on the right calf.
Acta Derm Venereol 2020; 100: XX–XX.
Diagnosis: Superficial angiomyxoma
Histopathology revealed a dome-shaped, otherwise unremarkable, epidermis. In the underlying dermis and subcutis a highly vascularized lobular tumour, consisting of spindle cells with only mild atypia, was evident (Fig. 2a). The tumour was accompanied by an infiltrate, mainly consisting of granulocytes and a myxoid stroma (Fig. 2b). Immunohistochemistry staining revealed positivity for CD34, vimentin and alpha-smooth muscle actin, whereas the spindle cells were negative for AE1/3, S-100 protein and desmin. Due to positive margins and the high risk of recurrence, re-excision with 5-mm safety margins was performed.
Apart from piebaldism, no further dermatological conditions were noted. Furthermore, there was no clinical evidence for Carney complex, specifically no pigmented skin lesions, and no cardiac myxomas or endocrine abnormalities. Sonography of the popliteal and inguinal lymph nodes was negative.
Superficial angiomyxoma (SA) was first described by Allen et al. (1) in 1988 and later characterized further histopathologically by Calonje et al. (2). SA is a rare (fewer than 150 reported cases in the literature) benign cutaneous tumour that usually presents as a slow-growing nodular or polypoid lesion. SA often invades the superficial and deep dermal layers, but does not invade the deep subcutaneous layers, in contrast to aggressive (deep) angiomyxoma, which is a rare mesenchymal neoplasm of the pelvic and genital regions identified almost exclusively in adult women (3). SA usually has a predilection for the trunk, the head and neck area, as well as the lower extremities (1). Congenital SAs are rare; however, congenital lesions presenting as parotid tumour mimicking angiomyxoma as well as congenital scalp lesions have been reported.
SAs have a recurrence rate of 30–40%, but no meta-static potential (2, 4). Differential diagnoses include focal cutaneous mucinosis, mucoid pseudocyst, neurothekeoma, myxoid neurofibroma, low-grade fibromyxoid sarcoma, low-grade myxofibrosarcoma, superficial acral fibromyxoma, and myxoid dermatofibrosarcoma protuberans.
The aetiology of SA formation is complex and not yet fully understood. A disruption of the lymphatic network with a possible obstruction of the trafficking of immune cells has been described (5).
This is the first described case of SA in an immunosuppressed patient. Interestingly, Zhu et al. has previously described a case of a large SA of the vulva complicated with condyloma acuminatum and Staphylococcus hominis infection due to an immunocompromised area after laser treatment (5). SAs in association with Carney’s complex (CC) were first described in 1985 (6). CC is a rare autosomal dominant disorder characterized by multiple myxomas, pigmentary abnormalities (e.g. blue naevi, lentigines) and an endocrine hyperactivity. Histologically, SAs are mainly dermal lesions with frequent extension to the subcutis. SAs are composed of spindle-shaped to stellate cells in a myxoid stroma with a predominantly vascular component (7). Vimentin is usually positive, CD34 can be positive in SA. An epithelial component (keratin cysts, hair follicules, basaloid cell proliferations or even trichofolliculomas) is often present. Some authors consider this component a genuine element of angiomyxoma, others as entrapped structures (1, 8). Dermoscopic features of SA include red exophytic globules, known as “red plant sign” or dotted and short curved linear vessels on a red-pinkish background (9). Given the high recurrence rates, Mohs surgery represents an adequate therapeutic option for SAs in cosmetically or functionally sensitive anatomical regions, especially in the head and neck region (10).
In conclusion, non-pigmented slow-growing nodules with dotted vessels on dermoscopy should include angiomyxoma in the differential diagnosis. It is important to differentiate SA from other myxoid tumours. SAs have an overall good prognosis with no metastasis. Although it is a distinct entity, it is advisable to rule out an association with Carney complex.
Fig. 2. Histopathology. (a) Section showing a highly vascularized lobular tumour consisting of spindle cells with only mild atypia (haematoxylin and eosin (HE) stain, original magnification ×25). (b) Section showing a granulocytic infiltrate within a myxoid stroma (HE stain, original magnification ×200).


 Click to show fullsize
Click to show fullsize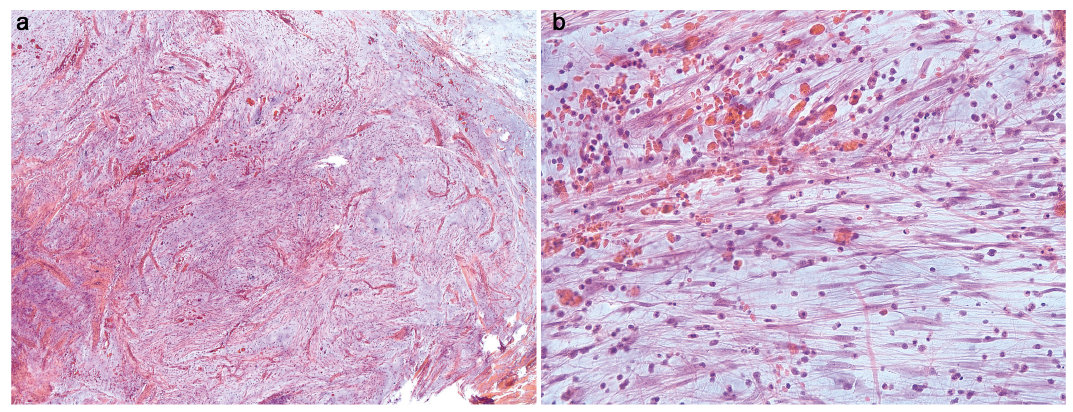 Click to show fullsize
Click to show fullsize