


Center for Human Genetics, KU Leuven, Leuven, Belgium
Neurofibromatosis type 1 (NF1) is the most common disorder characterized by multiple café-au-lait macules. Most individuals with this autosomal dominant disorder also have other features, such as skinfold freckling, iris Lisch nodules and benign or malignant peripheral nerve sheath tumours. Legius syndrome is a less frequent autosomal dominant disorder with similar multiple café-au-lait macules and skinfold freckling. Legius syndrome is not characterized by an increased risk of tumours, and a correct diagnosis is important. In young children with a sporadic form of multiple café-au-lait macules with or without freckling and no other manifestations of NF1 these 2 conditions cannot be differentiated based on clinical examination. Molecular analysis of the NF1 and SPRED1 genes is usually needed to differentiate the 2 conditions. Other less frequent conditions with café-au-lait macules are Noonan syndrome with multiple lentigines, constitutional mismatch repair deficiency and McCune-Albright syndrome.
Key words: CAL; NF1; Legius syndrome; SPRED1.
Accepted Feb 12, 2020; Epub ahead of print Mar 9, 2020
Acta Derm Venereol 2020; 100: adv00093
Corr: Eric Legius, Centre for Human Genetics, Herestraat 49, BE-3000 Leuven, Belgium. E-mail: Eric.Legius@uzleuven.be
Neurofibromatosis type 1 and Legius syndrome are both autosomal hereditary conditions with the same type of hyperpigmentation macules and skinfold freckles. Patients with neurofibromatosis type 1 usually develop additional signs, such as tumours of the peripheral nerves, and iris Lisch nodules. At a young age these additional signs might not be present, and the correct diagnosis can only be made by genetic testing, because these 2 conditions are caused by mutations in different genes. A correct diagnosis is essential because the medical follow-up is different.
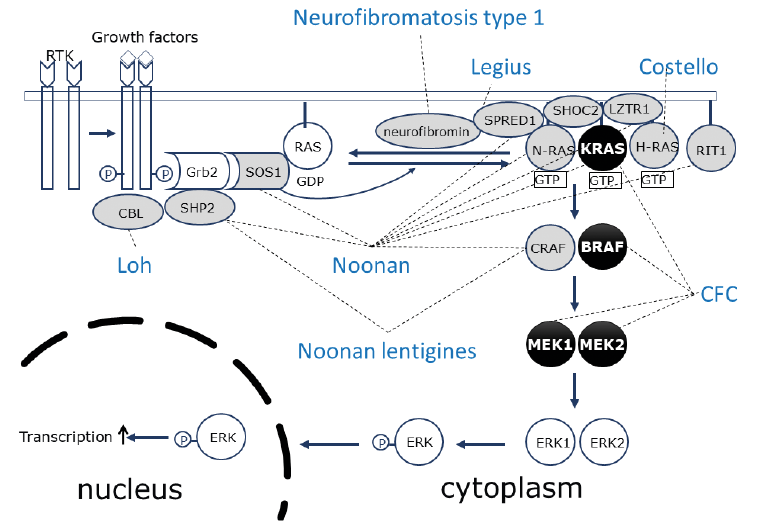
In 2007 an “NF1-like syndrome” was reported resulting from heterozygous mutations in the SPRED1 gene. The phenotype of affected patients in this autosomal dominant condition resembled the phenotype of neurofibromatosis type 1 (NF1) (1). More specifically, these patients show the same multiple café-au-lait macules (CALMs) typically seen in patients with NF1. Freckling in the axillary region and the groin is another feature that is equally present in both syndromes. Unlike the dermatological phenotype, other phenotypic features differ substantially between the 2 syndromes. This NF1-like syndrome is considered to be a milder condition than NF1, since neurofibromas and other typical tumoural manifestations of NF1 are not present. In order to differentiate clearly between both disorders the NF1-like syndrome was named “Legius syndrome” at the 13th European Neurofibromatosis Meeting (2008). Both NF1 and Legius syndrome are caused by mutations in genes related to the rat-sarcoma-mitogen-activated protein kinase (RAS-MAPK) signalling pathway. This review summarizes overlapping and non-overlapping clinical features of these 2 syndromes, as well as their underlying molecular mechanism and relationship with other disorders caused by mutations in the same signalling pathway (Fig. 1) (the so-called RASopathies).
Fig. 1. The RAS-MAPKinase pathway and the proteins involved in the different RASopathies. SOS: son of sevenless; RAS: rat sarcoma; SHOC2: suppressor of clear homolog; LZTR1: leucine zipper like transcription regulator 1; N-RAS: neuroblastoma RAS; KRAS: Kirsten RAS; H-RAS: Harvey RAS; RIT1: RAS like without CAAX 1.
The clinical phenotype of NF1 is characterized by multiple CALMs, skin-fold freckling and Lisch nodules in the iris. Patients with NF1 need clinical surveillance during childhood because of the risk of multiple complications, such as optic pathway gliomas, learning difficulties, social and emotional difficulties, skeletal problems, such as scoliosis and tibial pseudarthrosis and disturbances in growth (2, 3). Patients with NF1 have a high risk of development of neurofibromas. Neurofibromas are benign nerve sheath tumours composed of different cell types. The tumoural cells in the nerve sheath tumours are the Schwann cells. Cutaneous neurofibromas are benign and mostly start appearing at puberty and continue to arise in adulthood. Their number and size can increase with age. Plexiform neurofibromas are frequently diagnosed in early infancy and can grow throughout childhood. During adulthood their growth tends to stabilize. They can be asymptomatic, although they can also cause pain and disfigurement. Internal plexiform neurofibromas cannot be discovered by clinical examination alone. Nodular plexiform neurofibromas that continue to grow in adulthood are at risk of malignant degeneration. They might evolve into an atypical neurofibroma and further progress to a high-grade malignant peripheral nerve sheath tumour (MPNST). Adults with NF1 should be monitored during their lifetime for abnormal growth of plexiform neurofibromas, as well as for the appearance of some other tumours, such as pheochromocytoma, glomus tumours of the digits, gastrointestinal stromal tumours and breast cancer in females (4). The phenotype in patients with NF1 can be extremely variable, even within families.
Inactivating mutations in the NF1 gene were identified as the molecular cause for NF1 in 1990 (5–7). In half of patients a de novo NF1 mutation is identified, and in the other half the mutation is inherited from one of the parents. Most mutations identified are limited to the NF1 gene, but approximately 5% of individuals have a microdeletion on chromosome 17q11.2 including the NF1 region and other genes. These patients have a more severe tumoural phenotype with more neurofibromas at a younger age and a 2-fold increased risk of MPNST. Moreover, these patients sometimes present with an overgrowth phenotype and usually have more learning problems and a lower mean total IQ score compared with individuals with intragenic NF1 mutations (8, 9). A milder phenotype, consisting of CALM and skinfold freckling, but without neurofibromas, is seen in individuals with a 3-bp in-frame deletion of exon 17 (c.2970-2972 delAAT) (10, 11) and in patients with a missense mutation at codon 1809 (12). NF1 individuals with a missense mutation affecting codons 844 to 848 generally show an important internal neurofibroma load (13).
Diagnostic criteria for NF1 were established at the National Institutes of Health Consensus Development Conference in 1988. However, young children with-out a family history of NF1 frequently do not fulfil the diagnostic criteria, because they often only show multiple CALMs. The other diagnostic criteria of the disease are frequently seen only later in childhood or adulthood. Moreover, differential diagnosis with other CALM-manifesting disorders is often difficult on clinical grounds. Since molecular techniques for identifying the underlying genetic mutation have become increasingly available, molecular genetic testing is now performed more frequently at initial diagnosis in order to differentiate from other CALM-presenting disorders and to guide clinical follow-up.
The NF1 gene is a tumour suppressor gene, and NF1-associated tumours show a bi-allelic inactivation of NF1 (14). A somatic inactivation of the wild-type NF1 allele is needed in combination with the germline NF1 mutation in a specific cell to start the oncogenic process. In neurofibromas a second hit is found in the Schwann cells, and they have been identified as the cells driving the growth of the neurofibromas (15, 16).
NF1 codes for the neurofibromin protein, which is highly conserved among species and is composed of different domains. The RAS-GTPase (guanosine triphosphatase) activating protein (GAP)-related domain (NF1-GRD) is the best-studied functional domain of the NF1 gene and corresponds to a small region located in the central part of the protein. GAP proteins are negative regulators of rat sarcoma (RAS); they stimulate the hydrolysis of RAS-GTP to RAS-GDP, converting RAS from the active to the inactive form. This NF1-GRD interacts with active RAS through an arginine finger of neurofibromin that binds to RAS in a specific groove. This interaction results in a GAP-stimulated hydrolysis of GTP (17). Inactivating mutations in the NF1 gene result in an increased activation of the RAS-MAPK pathway due to a deficient downregulation of active RAS proteins.
NF1 and Legius syndrome are part of a group of overlapping disorders previously known as the Neuro-Cardio-Facio-Cutaneous (NCFC) syndromes (18). The phenotype associated with this group of disorders consists of neurological symptoms (e.g. psychomotor delay, learning difficulties, intellectual disability), cardiac abnormalities (most frequently pulmonary valve stenosis and hypertrophic cardiomyopathy), facial features (e.g. hypertelorism, ptosis, low implanted posteriorly rotated ears) and cutaneous findings (e.g. café-au-lait macules). Other frequently encountered features in these conditions were short stature and macrocephaly. Moreover, an increased risk of malignancy has been described in some of these syndromes. This group of disorders consists of NF1, Costello syndrome, cardio-facio-cutaneous syndrome, Noonan syndrome, Noonan syndrome with multiple lentigines, Loh syndrome, and Legius syndrome. These disorders not only share phenotypic features, but they are also caused by mutations in genes coding for proteins of the RAS-MAPK pathway. These disorders are now grouped under the name RAS-MAPK syndromes or RASopathies. A review of RASopathies can be found in (19).
This RAS-MAPK pathway had previously been extensively studied for its role in cancer biology. RAS genes are proto-oncogenes controlling pathways that are important regulators of cell growth. Many solid tumours show mutations in one of the RAS genes. The RAS homologues (neuroblastoma RAS (NRAS), Kirsten RAS (KRAS), Harvey RAS (HRAS)) code for proteins that are active in the GTP-bound and inactive in the GDP-bound state. Membrane-bound receptor tyrosine kinases are activated by binding to growth factors, and this leads via different adaptor proteins to activation of RAS-guanosine nucleotide exchange factors (GEFs) such as son of sevenless (SOS). RAS-GEFs activate RAS by stimulating the exchange of GDP to GTP bound to RAS. Active RAS-GTP has different downstream effector molecules. GTP-bound RAS binds to and actives the serine-threonine kinase rapidly accelerated fibrosarcoma (RAF) (MAPKKK= MAPkinasekinasekinase). Activated RAF-kinases phosphorylate and activate the protein kinase MEK (MAPKK= MAPKinasekinase). Active MAPK-ERK kinase (MEK) kinases (MEK1 and MEK2) phosphorylate a threonine and tyrosine on their only known substrate MAPKinase (ERK) (MAPK= MAPkinase). ERK activates transcription factors and signalling proteins. Activation of the RAS-MAPK signalling cascade thus results in stimulation of cell proliferation, promotion of cell survival and control of cell differentiation. Signalling is downregulated when RAS-GTP is hydrolysed to RAS-GDP. RAS proteins have intrinsic GTP-ase activity, which is strongly stimulated by GTP-ase activating proteins (GAPs), such as neurofibromin.
Some rare large families with autosomal dominant Noonan syndrome showed linkage to a locus on chromosome 12q24.1. Later it was shown that activating mutations in tyrosine-protein phosphatase non-receptor type 11 (PTPN11), located in this region on chromosome 12, were identified for a large group of Noonan syndrome individuals. PTPN11 codes for the Src homology region 2 (SH2)-containing protein tyrosine phosphatase (SHP2) protein, which interacts in a stimulating way with the RAS signalling cascade (19). Noonan syndrome is an autosomal dominant syndrome characterized by short stature, a specific facial dysmorphism, macrocephaly, ptosis of the eyelids, epicanthal folds, low implanted and posteriorly rotated ears, low posterior hairline and a broad webbed neck. Widely spaced nipples and pectus abnormalities are also frequently observed, but are less specific. Heart defects, such as pulmonic stenosis and hypertrophic cardiomyopathy, are found in 50–80% of patients. Developmental delay can be present and is rather mild.
Heterozygous mutations in HRAS were identified in individuals with Costello syndrome in 2005 by Aoki et al. (20, 21). This was a remarkable finding, because it was the first time that constitutional mutations in one of the RAS genes was identified in a human disorder. Prior to that report it was assumed that germline dominant activating mutations in one of the RAS genes were not compatible with life. Costello syndrome is a sporadic disorder. It usually presents with high birthweight and neonatal feeding problems. Postnatal failure to thrive and growth retardation are observed. Patients with Costello syndrome have redundant subcutaneous tissue with deep palmar and plantar creases. Coarse facial features and cardiac abnormalities are frequent. Relative macrocephaly and intellectual disability are usually present. Many individuals develop papillomata in the peri-oral and peri-anal region. Tumour risk by 20 years of age is estimated at 15% and rhabdomyosarcoma, neuroblastoma and bladder carcinomas are observed.
Knowledge of the genetic mechanisms in this group of disorders has been expanding rapidly over the years. Mutations in several other genes of the RAS-MAPK pathway were identified in Noonan syndrome (KRAS, NRAS, SOS1, BRAF, RAF1, suppressor of clear homolog (SHOC2), RAS like without CAAX 1 (RIT1), Casitas B-lineage lymphoma (CBL) and eucine zipper like rranscription regulator 1 (LZTR1)) and in cardio-facio-cutaneous syndrome (CFC) (BRAF, MEK1, MEK2 and KRAS). Germline KRAS mutations do not overlap with the mutational hotspots in solid tumours. KRAS is an important protein during embryogenesis. Strongly activating mutations in KRAS as seen in cancer tissues are most probably not tolerated in the germline and are probably lethal during development.
Linkage analysis in 2 families with multiple CALMs and freckling, but without a pathogenic NF1 mutation was used to map the condition to a region on chromosome 15 where SPRED1 was localized. Existing literature data at that moment pointed to the SPRED1 protein as a negative regulator of the RAS-MAPK signalling pathway (22). Sequencing of the SPRED1 gene in affected patients from those families showed inactivating heterozygous germline mutations in the SPRED1 gene as well as in 3 other families and in 6 unrelated patients with a phenotype of “familial CALM only” (1).
The Spred1 gene (Sprouty-related, EVH1 domain containing 1) was identified in 2001 and has 7 exons coding for 444 amino acids. The SPRED1 protein has 3 functional domains: an N-terminal EVH1-domain, a central c-KIT-binding domain and a C-terminal SPRY-related domain. The highest expression of human SPRED1 is seen in lung, brain, spinal cord and spleen. Expression is lower in liver, pancreas, muscle, prostate, heart, thymus, kidney and bone marrow.
The initial report by Brems et al. reported families with a phenotype similar to the phenotype seen in mild cases of NF1, showing multiple CALMs, axillary freckling, macrocephaly and sometimes Noonan-like facial features. Learning difficulties and/or attention deficits were less frequent compared with NF1. Of special note is the observation of multiple lipomas in several adults in 2 unrelated families. Some typical features of NF1 were not observed, such as Lisch nodules, typical bone defects, and NF1-associated tumours (1).
After this first report SPRED1 mutation analysis in several other cohorts of patients in follow-up in a multidisciplinary outpatient clinic for patients with NF1 were reported. Pasmant et al. (23) identified 5 unrelated individuals with a SPRED1 mutation in 61 cases. They confirmed the phenotype observed in the first publication with CALMs, freckling and learning disability without neurofibromas or Lisch nodules. Lipomas were seen in only one family.
In another study 6 individuals were identified with SPRED1 mutations in 85 unrelated patients negative for an NF1mutation. None of the 6 had cutaneous neurofibromas and 5 out of 6 individuals met NF1 diagnostic criteria (24). All individuals had multiple CALMs. Noonan-like facial features, macrocephaly, Lisch nodules or neurofibromas were not reported, and developmental or learning problems were not described.
Messiaen et al. (25) reported a genotype-phenotype study in 22 unrelated individuals carrying a SPRED1 mutation. These 22 individuals were identified through clinical testing. Fifty percent fulfilled the NIH diagnostic criteria for NF1due to multiple CALMs with or without freckling and/or a positive family history. No increased frequency of lipomas was reported. Other NF1 diagnostic features, such as symptomatic optic pathway gliomas, neurofibromas or osseous lesions, were not present. Relative macrocephaly was observed in 27% and language/speech problems were mentioned in 25% of children. In a separate cross-sectional study SPRED1 mutation analysis was performed in 1,318 unrelated patients with a NF1 phenotype but without a NF1 mutation (25). In 33 unrelated individuals 26 different pathogenic SPRED1 mutations were identified. Seven, probably benign, missense mutations were seen in 9 individuals. In 19% of NF1 mutation-negative families with an autosomal dominant phenotype of “CALMs only” with or without freckling a pathogenic SPRED1 mutation was detected. Following this study, it can be estimated that 1–4% of individuals with multiple CALM have Legius syndrome (26, 27).
In a report on individuals from 14 families with Legius syndrome one patient had a vestibular schwannoma and one a desmoid tumour. It is not known whether these tumours are related to the germline SPRED1 mutation. (28). Learning difficulties were observed in 14/25 individuals. Unilateral postaxial polydactyly was found in 2 patients in this study and in one patient reported by Messiaen et al. (25).
A small study investigated whether Legius syndrome is associated with neurocognitive problems, since learning difficulties (1, 23, 29), hyperactivity (1, 25) and language or speech delay (23, 25) had been reported in Legius syndrome and other RASopathies are also associated with neurocognitive problems (30). In 15 patients with Legius syndrome a mean Full scale intelligence quotient (FSIQ) of 101.57 (SD=17.57; median=107; IQR=23) was reported, which did not differ significantly from the control group (unaffected siblings). The FSIQ was higher than the mean FSIQ in 103 patients with NF1 from the same outpatient clinic. These preliminary data suggest that, in addition to the somatic phenotype (25), the cognitive phenotype is also milder in Legius syndrome than in NF1 and other RASopathies. In Legius syndrome individuals a large variability in mean FSIQ was observed. In comparison with NF1, there were few behavioural problems as assessed by the CBCL.
Another common feature of the RASopathies is the increased malignancy risk. This risk varies between the different RASopathies. It is low in individuals with Noonan syndrome, with an estimated 4% increase in cancer risk vs. a higher risk in Costello syndrome, estimated at 15% by age 20 years (31). In NF1 benign neurofibromas are seen in the majority of patients at an adult age. In children with NF1 the risk of an optic pathway glioma is estimated at 15%, but more than 2/3 are asymptomatic. The lifetime risk of MPNST is estimated at 10–15%, with a higher risk in patients with NF1 microdeletion. Adult women with NF1 have an increased risk of breast cancer between the ages of 30 and 50 years, and it is recommended to start screening at the age of 30 years for early detection of breast cancer (32, 33).
At present we cannot completely exclude that Legius syndrome is associated with an increased risk of malignancies. Pasmant et al. (34) found one leukaemia in a patient with Legius syndrome with a SPRED1 loss of heterozygosity in the leukaemic cells in a set of 230 paediatric lymphoblastic and acute myeloblastic leukaemias. Currently there is no documented increased risk of malignancies in Legius syndrome.
The CALMs in patients with NF1 and Legius syndrome are clinically indistinguishable. Naevus anaemicus has been suggested to be a clinical sign useful to differentiate NF1 from other CALM disorders (35). However, naevus anaemicus has been reported in a patient with Legius syndrome, as well as in a patient with Noonan syndrome with multiple lentigines due to a PTPN11 mutation. Naevus anaemicus is not specific for NF1 and cannot be used as a criterion to differentiate between NF1 and Legius syndrome (36).
Sporadic cases of CALMs without NF1 mutation are infrequently associated with a SPRED1 mutation and might represent NF1 mosaicism or other conditions (37). A specific surveillance for tumoural complications is not recommended in children and adults with Legius syndrome, in contrast to NF1.
It has been shown that SPRED1 binds to neurofibromin with its EVH1 domain and it recruits neurofibromin to the plasma membrane. SPRED1 is anchored in the plasma membrane by its sprouty-related domain. At the plasma membrane SPRED1, neurofibromin and RAS form a multiprotein complex resulting in down-regulation of RAS-GTP levels (38).
Previously reported mutations and polymorphisms in the SPRED1 gene can be found in the Leiden Open Variation Database (http://www.lovd.nl/SPRED1). No clear mutational hotspots in the gene have been identified. Most of the pathogenic mutations are predicted to be truncating (nonsense or frameshift mutations). A minority are missense variants. Most of the missense variants are classified as benign polymorphisms. For some missense mutations functional characterization was able to classify them as pathogenic. In Legius syndrome cultured melanocytes from a CALM showed a biallelic mutation in the SPRED1 gene. The same mechanism (biallelic NF1 inactivation) was previously reported in melanocytes from CALM in NF1 (1).
Constitutive Mismatch Repair Deficiency (CMMRD) is an autosomal recessive inherited condition caused by bi-allelic mutations in the mismatch repair genes MLH1, MSH2, MSH6 or PMS2. The proteins encoded by these genes are responsible for correcting base substitution mismatches or insertion-deletion mismatches generated during DNA replication.
Heterozygous mutations in these genes are responsible for the autosomal dominant Lynch syndrome, a cancer predisposition syndrome characterized by increased risk of adult malignancy, including colorectal cancer, gynaecological cancer (ovarian cancer and endometrial cancer) and uro-endothelial tumours. Tumours in individuals with Lynch syndrome frequently demonstrate microsatellite instability (MSI) and lack of expression of the mutated MMR gene by immunohistochemistry.
Most frequently described malignancies in children with CMMRD are haematological malignancies, brain tumours and gastro-intestinal cancers, but also low-grade gliomas and premalignant gastro-intestinal lesions have been identified. These children present with multiple CALMs that are clinically difficult to distinguish from those in NF1 or Legius syndrome. A study from the international CMMRD consortium (39), showed cutaneous findings resembling NF1 in all children, suggesting that CMMRD should be considered in the differential diagnosis of children presenting with CALMs and other variables associated with CMMRD, such as consanguinity in the parents or a family history of childhood, brain, haematological or gastro-intestinal malignancies. In the review of Wimmer et al. (40), more than 60% (91/146) of the patients with CMMRD were reported to show at least 1 CALM or hyperpigmented skin area and 27/146 presented CALM and other signs of NF1. Interestingly, in up to 75% of families with CMMRD no Lynch-associated malignancies were identified in adult family members carrying the heterozygous MMR gene mutation (37). This is probably related to the fact that, in CMMRD pedigrees, mutations in PMS2 and MSH6 are mostly found. These genes are known to be less penetrant than the other Lynch syndrome-associated genes. Diagnostic criteria for CMMRD are given in a review paper by the C4CMMRD consortium (40).
CALMs can also be found in other autosomal dominant conditions, including piebaldism, neurofibromatosis type 2 (NF2), Schwannomatosis, Noonan syndrome with multiple lentigines, and in McCune-Albright syndrome caused by mosaic mutations in the guanine nucleotide binding protein (G protein), alpha stimulating activity polypeptide 1 (GNAS) gene. The cutaneous phenotype in these latter conditions is often distinguishable from NF1 for trained clinicians.
Piebaldism is a rare autosomal dominant condition that is characterized by depigmented areas of the skin and hair. Patients often have a white forelock of hair and depigmented skin patches in a specific pattern. Irregularly shaped depigmented spots can be present on the face, trunk and extremities. Typical CAL spots can be present. The condition is caused by heterozygous mutations in the v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (KIT) proto-oncogene or sometimes in the zinc finger transcription factor snail family transcriptional repressor 2 (SNAI2).
NF2 is an autosomal dominant condition caused by mutations in the NF2 gene on chromosome 22. NF2 individuals develop typically bilateral vestibular schwannomas. Schwannomas localized on other nerves are also seen as well as meningiomas and ependymomas. Mononeuropathy occurring in childhood may present as facial nerve palsy or hand/foot drop. Multiple CALMs can be present in children with NF2, although usually there are fewer spots and they are smaller than in NF1. Moreover, they tend to be paler and have more irregular borders than in NF1. Hypopigmented areas can also occur (41).
A related disorder is familial Schwannomatosis, a rare autosomal dominant condition characterized by multiple schwannomas, predominantly occurring in the spine, but also in the peripheral nerves and cranial nerves. Heterozygous germline mutations in /SNF related, matrix associated, actin dependent regulator of chromatin, subfamily B, member 1 (SMARCB1) or LZTR1 have been reported is most individuals with familial Schwannomatosis, both located on chromosome band 22q11. Merker et al. (42) reported that 23% of patients had at least 1 CALM >1.5 cm; none had more than 4 CALMs > 1.5 cm. No intertriginous freckling was reported in these patients.
Noonan syndrome with multiple lentigines belongs to the group of RASopathies. This condition presents with a Noonan syndrome phenotype and multiple lentigines. The associated heart defect is frequently a hypertrophic cardiomyopathy or pulmonic stenosis. Sensorineural hearing loss is present in approximately 20% of patients and intellectual disability, usually mild in 30%. The condition can be caused by heterozygous mutations in BRAF, MEK1, PTPN11 or RAF1. A couple of CALMs are observed in a large number of patients and may precede the appearance of the typical lentigines, leading to a suspicion of NF1 or Legius syndrome in young children (43).
In individuals with fibrous dysplasia/McCune Albright syndrome (FD/MAS) large CALMs with irregular borders are seen in combination with polyostotic fibrous dysplasia. The large CALMs do not cross the midline. FD/MAS results from a postzygotic somatic activating mutation of GNAS. Characteristic features of CALMs in this condition are the irregular borders resembling the “coast of Maine” (in contrast to the smooth-bordered “coast of California” lesions seen in NF1) and the distribution which reflects the embryonic cell migration of melanocytes. Fibrous dysplasia (FD) can range from a monostotic lesion to severe polyostotic disease. Endocrinological complications can include gonadotropin-independent precocious puberty, thyroid abnormalities and growth hormone excess.
Legius syndrome and NF1 share a similar dermatological phenotype, consisting of multiple CALMs and freckling. Legius syndrome is a much milder condition lacking the tumour phenotype seen in NF1. The neurocognitive phenotype also seems milder. Since the number of reported patients is still limited it is uncertain whether some rare malignancies are associated with Legius syndrome, such as certain types of leukaemia. CALMs are the most frequent and easily recognizable manifestation of both conditions. In young children without other manifestations of NF1, differential diagnosis between the 2 conditions can be difficult on clinical grounds. Molecular genetic testing may help in establishing a correct diagnosis and ensure appropriate surveillance for the affected individuals. Another condition to consider in children with multiple CALMs is CMMRD. Although rare, it is important to recognize this syndrome because it is associated with a high risk of childhood malignancies. CMMRD should be considered in children with CALMs from consanguineous parents or with a personal or family history of childhood haematological malignancies, brain tumours, gastro-intestinal malignancies or pilomatricomas (40). A family history compatible with Lynch syndrome may be present, but is often lacking. Other CALM manifesting disorders can usually be distinguished by their disease-specific manifestations and different aspect of the CALMs.


 Click to show fullsize
Click to show fullsize