


1Institute of Human Genetics, Medical Center, University of Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany, and 2Department of Dermatology, Reference Center for Rare Skin Diseases MAGEC, Saint Louis Hospital AP-HP, Paris, France
Inherited ichthyoses are classified as Mendelian disorders of cornification (MEDOC), which are defined on the basis of clinical and genetic features and are mainly divided into non-syndromic and syndromic ichthyoses. Numerous genes, which encode for corresponding proteins, are involved in the normal differentiation of keratinocytes (cornification) and participate in the formation of a functional epidermal barrier. To date, mutations in more than 50 genes are known to result in various types of ichthyoses. Thanks to modern genetic diagnostic methods based on targeted next generation sequencing (NGS), approximately 80–90% of cases can be resolved at present. Further sequencing methods covering the whole exome (WES) or whole genome (WGS) will obviously elucidate another portion of the remaining unknown ichthyoses in the future.
Key words: Mendelian disorders of cornification; ichthyoses; ARCI; genes; mutations; molecular genetic diagnostics.
Accepted Feb 12, 2020; Epub ahead of print Mar 9, 2020
Acta Derm Venereol 2020; 100: adv00096.
Corr: Judith Fischer, Institute of Human Genetics, Medical Center, University of Freiburg, Faculty of Medicine, University of Freiburg, Breisacher Str. 33, DE-79106 Freiburg, Germany. E-mail: judith.fischer@uniklinik-freiburg.de
Knowledge of the molecular genetic causes and mechanisms of hereditary ichthyoses has increased hugely since the 1990s due to the ubiquitous application of modern sequencing technologies. It is important for doctors and scientists that this new knowledge is clinically and genetically correctly classified, in order to make diagnosis and differential diagnosis easier. This article provides an overview of the genetic background and clinical features of ichthyoses and related cornification disorders.
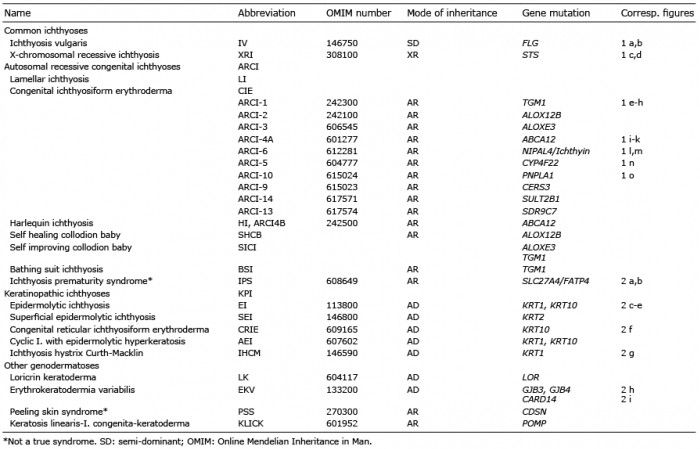
Ichthyoses are genetically determined monogenic (Mendelian) cornification disorders of the epidermis characterized by different degrees of scaling, hyperkeratosis and erythroderma, often associated with palmoplantar keratoderma (PPK) or hyperlinearity. Non-syndromic ich-thyoses are limited to skin symptoms and can be subdivided into common and rare forms (Table I), whereas syndromic forms are classified according to the additional symptoms (Table II).
Table I. Non-syndromic ichthyoses
Table II. Syndromic ichthyoses
In congenital ichthyoses, the skin symptoms are present at birth, either as collodion membrane (CM) or as congenital ichthyosiform erythroderma (CIE). Collodion babies (CB) later develop a lamellar ichthyosis (LI) or CIE, or the rarer variants of self-improving collodion ichthyosis (SICI) or bathing suit ichthyosis (BSI) (1).
In common ichthyoses, such as ichthyosis vulgaris (IV) and X-linked recessive ichthyosis (XRI), skin manifestations do not appear until several weeks to months after birth. Occasionally, mild scaling may occur in patients with XRI at birth, which then initially regresses and usually begins again at the age of 4–6 months (1).
In addition to the 2 common forms, non-syndromic ichthyoses also include the much rarer autosomal recessive congenital ichthyosis (ARCI) that clinically manifests as harlequin ichthyosis (HI), LI, or CIE (2).
Ichthyoses caused by keratin mutations, such as epidermolytic ichthyosis (EI), superficial epidermolytic ichthyosis (SEI), and congenital reticular ichthyosiform erythroderma (CRIE), are referred to as keratinopathic ichthyoses. They manifest at birth and often feature episodes of blistering. Most of these types are inherited as autosomal dominant traits, but autosomal recessive forms have also been described on occasion (2).
The family history and pedigree survey can provide important conclusions about the mode of inheritance, and thus contribute to the correct diagnosis. Modern sequencing methods (e.g. next generation sequencing; NGS), including multi-gene-panel sequencing or whole-exome sequencing (WES), help to confirm the suspected diagnosis quickly and reliably.
The 2 most common types of ichthyosis are IV and XRI, whereas ARCI, keratinopathic ichthyosis and a few other non-syndromic forms are much rarer.
Ichthyosis vulgaris
The most common form of ichthyosis is IV, with a prevalence of up to 1:100 (3). It is caused by autosomal semi-dominant inherited loss-of-function mutations in the filaggrin gene (FLG). In the majority of patients (approximately 2/3) 2 FLG mutations can be detected (4), which are associated with a relatively severe phenotype, whereas patients with only one mutation are significantly more mildly affected. The IV is associated with atopic eczema in approximately half of cases and approximately 40% with allergic rhinitis, conjunctivitis or bronchial asthma, e.g. also overlapping with atopic eczema. Approximately one-third of patients have no atopy (4). Histological analysis reveals an orthohyperkeratosis (thickening of the stratum corneum) with simultaneously reduced or absent stratum granulosum. Electron microscopy shows the defect as reduced, very small (crumbly) keratohyalingranulae. The typical clinical picture of IV is characterized by a fine, pale-grey scaling (Fig. 1a, b) with the exception of the large articular flexures and a palmo-plantar hyperlinearity and keratosis pilaris.
Fig. 1. Examples of skin signs in non-syndromic ichthyoses. (a, b) Fine, pale-grey scales of ichthyosis vulgaris on thorax and legs of a patient with compound heterozygous filaggrin (FLG) gene mutations. (c, d) Adherent, rhomboid, dark-brown scaling in X-linked recessive ichthyosis; hands and feet are not affected. (e–h) Severe lamellar ichthyosis and thick palmoplantar keratoderma in a patient with autosomal recessive congenital ichthyosis (ARCI) due to homozygous mutations in TGM1. (i–k) Ichthyosiform erythroderma and severe palmoplantar keratoderma in patients with ABCA12 mutations. (l, m) Lamellar ichthyosis and yellow plantar keratoderma in patients with NIPAL4 mutations. (n) Typical palmar hyperlinearity in a patient with CYP4F22 mutations. (o) In patients with PNPLA1 mutations cyclic superficial scaling can be observed.
X-linked recessive ichthyosis
XRI is the second most common form of ichthyosis, with a prevalence of 1 in 2,000 boys (1). It is caused by steroid sulphatase (STS) deficiency (5) and is often associated with further clinical problems, such as cryptorchidism (~20%) or social communication deficits, such as attention deficit hyperactivity syndrome (40%) or autism (25%) (6). The majority of patients present deletions of a part or the totality of the STS gene (isolated non-syndromal XRI); only 10% of cases are due to point mutations. Larger deletions, which also spread to neighbouring genes, lead to much more complex diseases, such as Kallmann syndrome, which is additionally associated with mental retardation, hypogonadism and anosmia. These contiguous gene deletion syndromes are then classified as syndromal ichthyosis. XRI can also be confirmed enzymatically by the determination of sulphatase activity in the blood. The lack of cholesterol hydrolysis leads to the accumulation of cholesterol-3-sulphate in the epidermis. Histological analyses may reveal a normal or rather thickened stratum granulosum (light microscopy), and a lack of degradation of the corneodesmosomes (electron microscopy) as a sign of the retention hyperkeratosis. The predominantly adherent, rhomboid, light-grey to dark-brown scaling is extended over the entire body, with the exception of the hands, feet and the flexor sides of the elbows and knees (Fig. 1c, d). Mothers of affected boys are carriers, who frequently report complications during the birth of their children (weakness of labour) followed by caesarean section or forceps birth.
Autosomal recessive congenital ichthyosis (ARCI)
The generic term ARCI refers to all non-syndromic forms of autosomal recessive congenital ichthyoses that are present at birth and not associated with blistering. This includes HI, which is by far the most severe form of ichthyosis, LI and CIE (2).
Prevalence studies in Germany and Spain show almost identical values of 1.6–1.7: 100,000 (7, 8). Histologically, the different ARCI types show typical signs of epidermal hyperproliferation with orthohyperkeratosis and thickened stratum granulosum, as well as signs of inflammation with lymphohistiocytic infiltrate of the dermis. Using electron microscopy (EM) it is sometimes possible to detect a change that is typical for the particular defect, e.g. cholesterol clefts in the stratum corneum in patients with TGM1 and PNPLA1 mutations, or inflated lamellar bodies in HI.
At present, mutations in 11 different genes are known to cause ARCI (see Table I):
Transglutaminase 1 TGM1 (ARCI1). The most common causes of ARCI are mutations in the TGM1 gene, first described in 1995 (9, 10) and found in approximately one-third of all cases of ARCI (11). The prevalence in Germany is given as 1:200,000 (7). Patients with TGM1 mutations are born in 80–90% of cases as a collodion baby and often present severe ectropion. The clinical picture is manifested in approximately 90% as LI and in approximately 10% as CIE (Fig. 1e–h). In general, there are no indications for a genotype-phenotype correlation. However, in some specific phenotypes, such as BSI or self-healing collodion baby, a correlation with specific mutations has been observed (12, 13).
Lipoxygenases ALOX12B (ARCI2) and ALOXE3 (ARCI3). Mutations in 1 of the 2 lipoxygenase genes ALOX12B or ALOXE3 were identified in 2002 using homozygosity mapping in consanguineous ARCI families (14). Overall, 17% of ARCIs are caused by mutations in 1 of the 2 lipoxygenase genes, with 12% ALOX12B and 5% ALOXE3 (11). The 2 enzymes 12R-LOX and eLOX3 catalyse the first 2 steps in the degradation pathway of arachidonic acid (15). Clinically, both LI and CIE occur. Especially in Scandinavian patients with ALOX12B mutations, a positive development of the phenotype towards self-improving collodion ichthyosis (SICI) is frequently observed (16, 17).
ATP-binding cassette transporter ABCA12 (ARCI4A and ARCI4B). Defects in the ABCA12 gene can either lead to LI (ARCI4A) or to a HI (ARCI4B), depending on the nature of the mutation (Fig. 1i–k). In 2003, homozygous missense mutations in ABCA12 were identified in patients from consanguineous North African families, leading to severe LI, hand and nail deformities, and kyphoscoliosis (18). In 2005, loss of function mutations in the same ABCA12 gene were identified as the molecular genetic cause of HI (19, 20). The life-threatening HI phenotype is characterized by massively thickened skin with impaired skin barrier function, infection and water loss, requiring intensive care treatment (19, 21). ABCA12 is a transmembrane lipid transporter acting at the lamellar granules (LG) and the cell membrane of keratinocytes. The ABCA12 transporter is important in delivering glucosylceramides (GluCer) to the lipid lamellae through lamellar bodies (LBs) (22).
NIPAL4 (ICHTHYIN) (ARCI6). In 2004, positional cloning was used to identify mutations in ICHTHYIN, which was later referred to as NIPAL4, according to official nomenclature (23). Approximately 16% of patients with ARCI have mutations in this gene (11), and the recurrent mutation p.Ala176Asp occurs in half of these patients. In some of the patients, a special phenotype is noted with typical reticular lamellar ichthyosis and pronounced palmoplantar keratoderma with central cut-outs (Fig.1 l, m). EM classifies these patients as type III with hyperkeratotic stratum corneum and stratum granulosum with vacuoles.
Cytochrome-P450 CYP4F22 (ARCI5). ARCI due to mutations in CYP4F22 occurs in 8% of cases and results in a relatively mild LI that may be accentuated in the periumbilical region. The patient is usually not born as a collodion baby and, similar to the IV, shows marked palmoplantar hyperlinearity (Fig. 1n) (24, 25).
Patatin-like phospholipase domain-containing protein 1 PNPLA1 (ARCI10). To identify mutations in the gene PNPLA1, a spontaneous dog model with golden retrievers with ichthyosis was used (26). Some of the patients with ichthyosis subsequently tested for the human PNPLA1 showed mutations in this gene. In contrast to the newborn puppies who showed no signs of ichthyosis at birth, all patients were born as collodion babies, and later developed LI. In some patients a phenotype with cyclic skin peeling has been observed (Fig. 1o) (27).
Ceramide synthase 3 CERS3 (ARCI9). In 2013 ceramide synthase 3 (CERS3) mutations were identified in patients with ARCI, and this gene encodes the protein responsible for the de novo synthesis of ceramides in the skin (28, 29). Mutations in CERS3 cause reduced formation of ultra-long-chain epidermis-specific ceramides, which leads to defective epidermal differentiation of the skin and thus to a disruption of the skin barrier. Clinically, LI dominates with palmoplantar hyperlinearity and hyperkeratosis. Histologically there is an acanthosis with significant thickening of the stratum granulosum in a normal horny layer. Immunofluorescence microscopy localized CERS3 between the stratum corneum and the stratum granulosum.
Sulphotransferase family 2b, member 1 SULT2B1 (ARCI 14). In 2017 Heinz et al. identified mutations in sulphotransferase family 2B member 1 (SULT2B1) in ARCI (30). Cytosolic sulphotransferases form a large family of enzymes that are involved in the synthesis and metabolism of several steroids in humans. The absence of cholesterol sulphate, a metabolite of SULT2B1, and an increased level of cholesterol, indicate a disturbed cholesterol metabolism of the skin upon loss-of-function mutation in SULT2B1. Mutation in SULT2B1 leads to an ARCI phenotype via increased proliferation of human keratinocytes, thickening of epithelial layers, and altered epidermal cholesterol metabolism (30).
Short-chain dehydrogenase/reductase family 9C, member 7 SDR9C7 (ARCI13). Mutations in the gene SDR9C7 were first described in 2016 in patients with congenital ichthyosis; they presented with large erythematous scales over the entire body, with hyperkeratosis of the elbows and knees, mostly associated with palmoplantar hyperkeratosis. The severity of skin lesions decreased with age, and the face and scalp were mostly not affected. Fungal skin infections including onychomycosis were observed frequently. Light microscopic analysis showed mild hypergranulosis and marked hyperkeratosis of the epidermis (31, 32). Hotz et al. reported 7 patients with SDR9C7 mutations, which also showed a relatively mild ichthyosis with generalized dry and scaly skin and mild or local erythema. With one exception, the patients were not born as collodion babies (33).
Fatty acid transport protein 4 SLC27A4 (IPS). Ichthyosis prematurity syndrome (IPS) due to mutations in SLC27A4 was initially classified as a syndromic ichthyosis, however the authors and others (34) propose to classify IPS under ARCI. Patients with IPS are typically born well before the calculated date of delivery and often require artificial ventilation due to neonatal asphyxia. The reason for this is the obstruction of the foetal bronchi by massively shed skin scales in the amniotic fluid. At birth an impressive verrucous hyperkeratosis is present, more prominent on the head, forehead and trunk, which heals quickly (35, 36). Subsequent to the critical neonatal phase, mild ichthyosis, atopy, fine hair, and a typical follicular keratosis pilaris are seen (Fig. 2a, b). IPS is inherited as an autosomal recessive trait and is caused by mutations in the gene SLC27A4, which codes for fatty acid transport protein 4 (FATP4) (37).
Fig. 2. Examples of skin signs in non-syndromic (continued from Fig. 1) and syndromic ichthyoses. (a, b) Follicular hyperkeratosis of the body and affected axilla in adults with ichthyosis prematurity syndrome (IPS). (c, d) Verrucous hyperkeratosis in epidermolytic ichthyosis (EI) of the abdomen and legs caused by heterozygous mutations in KRT1. (e) Hyperkeratosis, erythroderma and skin fragility in EI due to a heterozygous mutation in KTR10. (f) Congenital reticular ichthyosiform erythroderma (CRIE) with specific heterozygous mutation in KRT10; white spots (representing normal skin) appeared since the age of 4 years due to revertant mosaicism. (g) Extensive, spiky hyperkeratosis over the extensor surfaces of the lower extremities in ichthyosis hystrix type Curth-Macklin (KRT1). (h) Erythrokeratodermia variabilis (EKV) due to a heterozygous mutation in the GJB3 gene, also known as CX31. (i) EKV due to a heterozygous mutation in CARD14. (j) Ichthyosis linearis circumflexa (polycyclic serpiginous migratory plaques with double-edged scales) in Netherton’s syndrome caused by biallelic SPINK5 mutations. (k) Pronounced, dark pigmented ichthyosis on the neck in a patient with Sjögren-Larsson syndrome and mutations in the ALDH3A2 gene. (l) Mild, ichthyosiform erythroderma in Chanarin-Dorfman syndrome.
Keratinopathic ichthyosis
The term keratinopathic ichthyosis (KPI) summarizes the forms that are caused by mutations in keratin genes (2). Inheritance in this disease group is usually autosomal dominant, although exceptionally, an autosomal recessive pattern of inheritance can occur. Typically, an epidermolytic hyperkeratosis is discovered using light microscopy, while collapsed keratin aggregates can be found by EM. These so-called tonofilaments have clumped/aggregated around the cell nucleus and lost their attachment to the desmosomes.
There are 3 main types of KPI: epidermolytic ichthyosis, superficial epidermolytic ichthyosis and congenital reticular ichthyosiform erythroderma (see Table I):
Epidermolytic ichthyosis. EI has previously been referred to as bullous ichthyosis, bullous CIE type Brocq, epidermolytic hyperkeratosis, or ichthyosis exfoliativa. EI is caused by mutations in the KRT1 (Fig. 2c, d) or KRT10 (Fig. 2e) genes. At birth there is usually a non-ichthyosiform erythroderma, which may be associated with blistering, which is why the differential diagnosis is bullous epidermolysis. Following the initial phase of blistering in the first few months of life, hyperkeratosis then occurs (2). Patients with KRT1 mutations have very severe PPK compared with patients with KRT10 mutation.
Superficial epidermolytic ichthyosis. SEI was formerly called ichthyosis bullosa Siemens and is caused by mutations in the KRT2 gene. Clinically it resembles EI, but shows a milder disease course with more localized skin symptoms. Since delineating the phenotype between EI and SEI is not always possible, KRT2 should be analysed in all patients with KPI in whom no mutations in KRT1 or KRT10 have been found (2).
Congenital reticular ichthyosiform erythroderma. CRIE is caused by specific mutations in KRT10 (38). The clinical picture at birth is dominated by pronounced erythroderma. Palmoplantar blistering and large scaling occurs, similar to peeling skin syndrome. In the later course, lichenification is also observed. In early childhood between the ages of 3 and 10 years, the development of multiple, small white spots begins; these spots can increase in size to 2 cm, which led to the French term “ichtyose en confettis” (Fig. 2f). The mechanism is mitotic recombination (2). This phenomenon of revertant mosaicism is also found in other diseases, e.g. epidermolysis bullosa. The same mechanism has been reported in cases with mutations in KRT1.
Other keratinopathic ichthyoses. In addition to the 3 main types of KPI (EI, SEI and CIE) there are also rarer types, such as cyclic ichthyosis with annular epidermolytic hyperkeratosis (AEI, OMIM 607602) and ichthyosis hystrix Curth-Macklin type (Fig. 2g) (IHCM, OMIM 146590).
Other non-syndromal ichthyoses
Other genodermatoses among the group of non-syndromal ichthyoses are included as they are phenotypically predominantly characterized by ichthyosis. Examples are the autosomal dominant inherited loricrin keratoderma (OMIM 604117), erythrokeratoderma variabilis (OMIM 133200) (Fig. 2h, i), and the 2 autosomal recessive disorders peeling skin syndrome (OMIM 270300) and KLICK syndrome (keratosis linearis-ichthyosis congenita-keratoderma, OMIM 601952), both inadvertently called “syndrome”, even though they are devoid of any extracutaneous involvement (Table I).
The syndromic ichthyoses are generally very rare and are classified based on the mode of inheritance as X-linked or autosomal inherited ichthyosis syndromes and can be further subdivided according to the predominant symptoms (2).
X-linked syndromes
The first group includes the syndromal form of XRI, XR IFAP syndrome and X-linked dominant (XD) chondrodysplasia punctata 2 (see Table II).
Syndromic X-linked ichthyosis. While in patients with mutations or minor deletions of the STS gene an isolated, skin-only XRI is present, larger deletions on Xp22.3 often involve multiple adjacent genes, termed “contiguous gene deletion syndrome”. As mentioned above, the additional symptoms depend on the extent of the deletion and range from mental retardation, hypogonadotrophic hypogonadism and anosmia in Kallmann syndrome (KAL1, OMIM 308700) to dwarfism (ISS, OMIM 300582, SHOX, OMIM 312865) and ocular albinism (OA1, OMIM 300500) (39, 40).
IFAP syndrome. The IFAP syndrome describes the triad of ichthyosis follicularis, alopecia (atrichia) and photophobia. Less than 100 male patients have been reported in the literature. Female carriers can sometimes present minimal symptoms, such as an asymmetrical distribution of body hair, patchy alopecia or hyperkeratosis along the Blaschko lines. In addition to the absence of scalp hair, eyebrows and eyelashes, the complete atrichia of body hair is part of the full spectrum of IFAP syndrome in male patients. There is often a pronounced ichthyosis follicularis with spine-like outgrowths of the skin follicles. The progressive blindness due to ulceration, scarring and vascularization of the cornea is a known complication. The allelic variant BRESHEK syndrome has additional symptoms, such as brain abnormalities, mental retardation, ectodermal dysplasia, skeletal deformities, Hirschsprung’s disease, ear and eye abnormalities, cleft palate, cryptorchidism and renal dysplasia or kidney hypoplasia. The terminology distinguishes IFAP syndrome with or without BRESHEK syndrome. These 2 phenotypes and also keratosis follicularis spinulosa decalvans (OMIM 308800) (41) are caused by mutations in the MBTPS2 gene (42).
Conradi-Hünermann-Happle syndrome (CDPX2). Conradi-Hünermann-Happle syndrome is also known as chondrodysplasia punctata 2. It is one of the XD inherited disorders that are lethal in male foetuses. Exceptions are possible in postzygotic mosaics or male chromosome sets with an excess X chromosome, as in Klinefelter syndrome (XXY). The characteristic symptoms of female patients include asymmetrical bone anomalies, sectoral cataracts, and streaky skin changes following the Blaschko lines. The stippling chondrodysplasia punctata is visible as a lime splash in the X-ray picture until approximately the ninth month of life. In the first few weeks of life, there is a very inflammatory phenotype with pronounced feather-like scaling and hyperkeratosis, which subsequently turns into linearly arranged follicular atrophoderma (43). CDPX2 is caused by mutations in the EBP gene, which codes for a delta (8) -delta (7) sterol isomerase, also known as emopamil binding protein, involved in cholesterol metabolism (44).
Autosomal inherited syndromes
The second group includes all other syndromic cornification disorders, which follow autosomal recessive or dominant inheritance, and can be further subdivided according to the most characteristic extracutaneous manifestations: Hair anomalies; Neurological symptoms; Fatal disease progression, Other typical symptoms (see Table II).
H1; Netherton syndrome. The AR inherited Netherton syndrome is caused by mutations in the SPINK5 gene, which codes for the serine protease inhibitor LEKTI. The impaired function of LEKTI leads to inflammatory processes in the epidermis and to a pronounced barrier disorder of the skin. At birth, generalized ichthyosiform erythroderma and severe growth and developmental deficiency are present, in part due to diarrhoea, intestinal malabsorption, hypernatraemic dehydration and recurrent infections. The erythroderma may persist, or develop into an “ichthyosis linearis circumflexa Comèl”, which is characterized by polycyclic serpiginous migratory plaques with typical double-edged scales (Fig. 2j). The typical hair anomalies can be detected by light microscopy, but usually only after the newborn phase. Bamboo hair (trichorrhexis invaginata) is considered pathognomonic for NS; trichorrhexis nodosa and pili torti can sometimes be observed. The scalp hairs are brittle and barely grow, eyelashes and eyebrows are also affected. There is a strong tendency to atopy (asthma, allergic rhinitis, atopic dermatitis, food allergies, urticaria and angioedema), increased serum IgE and hypereosinophilia.
H2; Ichthyosis hypotrichosis syndrome. The AR inherited ich-thyosis hypotrichosis syndrome (IHS) is caused by mutations in the ST14 gene, which encodes serine protease matriptase (45). It is also listed in OMIM as ARCI11; however it should be classified as syndromic ichthyosis with hair defect. Clinically, in addition to congenital ichthyosis and hypotrichosis, hypohidrosis and follicular atrophoderma are also found. The existing hair appears curly and brittle; sometimes it is pili torti or pili bifurcati. Eyebrows and eyelashes are usually sparse. Photophobia, blepharitis and corneal clouding have also been described in individual patients. Light microscopy shows a pronounced acanthosis and a thickened stratum corneum in the epidermis with orthohyperkeratosis. Using electron microscopy persistent corneodesmosomes and lamellar body-like deposits can be found in the horny layer.
H3; Ichthyosis hypotrichosis sclerosing cholangitis syndrome. IHSC is another inherited AR syndrome with congenital ich-thyosis, hypotrichosis and additional sclerosing cholangitis. The synonyms NISCH syndrome (neonatal ichthyosis sclerosing cholangitis) and ILVASC syndrome (ichthyosis leukocyte vacuole alopecia sclerosing cholangitis syndrome) are also common. Liver involvement can provide an important clue to diagnosis, but its severity is highly variable. Individual patients with progressive hepatic insufficiency who needed liver transplantation have been described. Hypotrichosis of the scalp is often associated with scarring alopecia and thinning of eyelashes and eyebrows. Other symptoms, such as oligodontia, hypodontia and enamel hypoplasia, have also been reported (46). Genetic causes are mutations in the CLDN1 gene, which codes for Claudin-1, a protein of tight junctions.
H4; Trichothiodystrophy. The term trichothiodystrophy (TTD) is based on the characteristic of the disease anomalies with short, brittle hair, longitudinal splitting and reduced content of sulphur-containing amino acids. Typically, a so-called tiger tail pattern with light and dark bands can be detected in polarization light. TTDs are classified as DNA repair or transcription disorders, and subdivided into various forms with or without photosensitivity. The autosomal recessive forms with ichthyosis are caused by mutations in the genes ERCC2, ERCC3 and GTF2H5. The hair anomalies are associated with skin manifestations, such as congenital ichthyosis, photosensitivity and nail abnormalities, as well as neurological symptoms, developmental and growth disorders (47).
N1; Sjögren-Larsson syndrome. This AR syndrome was named after the Swedish authors (48) who first described it in 1957. It is clinically characterized by congenital ichthyosis, intellectual deficit with delayed speech development and spastic paresis. At birth there are sometimes only mild hyperkeratoses, which then develop into a pronounced, generalized, often heavily pigmented, dirty-brown ichthyosis with an accentuation in the articular folds, neck, trunk and extremities (Fig. 2k). The SLS is caused by mutations in the ALDH3A2 gene, which codes for a fatty aldehyde dehydrogenase (FALDH), which oxidizes long-chain aldehydes to fatty acids. Neurological symptoms, such as spastic diplegia or quadriplegia and seizures, may appear later, after early childhood. Many patients never learn to walk and are in long-term care throughout their lives. Life expectancy is reduced. Eye involvement with crystalline retinal inclusions, corneal opacity and macular degeneration, as well as photophobia and myopia, are observed (1).
N2; Refsum syndrome. This AR disorder, named after a Norwegian author, is characterized by increased phytanic acid concentration, which can be detected in plasma or urine. During the course of the disease, specific damage to the retina, brain and peripheral nervous system occur. The symptoms are not present at birth, but usually occur after the age of 15 years. Night blindness (nyctalopia) is a typical first manifestation, followed later by neurological symptoms, such as distal motor polyneuropathy, cerebellar ataxia, mental retardation, deafness and anosmia. Ichthyosis only develops later in the course of the disease. Other symptoms, such as epiphyseal dysplasia, cardiomyopathy and retinitis pigmentosa, have also been reported. Refsum syndrome is an autosomal recessive disorder. In more than 90% of cases, mutations in the PHYH/PAXH gene coding for phytanoyl-CoA-hydroxylase can be detected. The function of the peroxisomal enzyme is the degradation of phytanic acid via α-oxidation. Less than 10% of mutations are found in the PEX7 gene (49).
N3; MEDNIK syndrome. An acronym for the symptoms of mental retardation, enteropathy, deafness, neuropathy, ichthyosis and keratoderma (50). This rare and severe AR multisystem disease is clinically and biochemically related to Menkes syndrome and Wilson disease, in which there is an accumulation of copper in the liver that can be treated with zinc acetate. The causes of MEDNIK syndrome are mutations in the AP1S1 gene, which codes for the σ1A subunit of the adapter protein complex 1 and controls the intracellular transport of the copper pumps ATP7A and ATP7B (51).
Recently, a new AR inherited syndrome has been described, showing mainly ichthyosis, deafness and photophobia. It is caused by mutations in the AP1B1 gene, which codes for the 1B-subunit of the adapter protein complex 1. There are some overlapping clinical features with MEDNIK syndrome; how-ever, the new AP1B1-syndrome seems to be less severe: the 5 described patients and our own case do not present neurological symptoms and have no or less -important enteropathy (52, 53).
N4; IKSHD syndrome. Heterozygous mutations in the ELOVL1 gene have been described in a new phenotype, in which, in addition to symptoms of the epidermis (ichthyosis, keratoderma) and the nervous system (spasticity, hypomyelination), dysmorphism (IKSHD) also occurs (54). So far, only 2 patients have been described in the literature, 1 of which certainly carried a neo mutation (54, 55). Similar to ELOVL4, ELOVL1 has functions in the elongation of fatty acids and is regulated by CERS2, a ceramide synthase, which is important for C24 sphingolipid synthesis (54). A previously described mouse model with Elovl1 deficit clinically showed a skin phenotype, and macroscopically reduced lipid lamellae and defective lamellar bodies in the stratum corneum (56).
F1; CEDNIK syndrome. The acronym CEDNIK syndrome derives from the typical constellation of symptoms, including cerebral dysgenesis, neuropathy, ichthyosis and palmoplantar keratoderma (cerebral dysgenesis, neuropathy, ichthyosis, palmoplantar keratoderma). This rare AR inherited neurocutaneous disease is caused by mutations in the SNAP29 gene and is characterized by severe developmental disorders of the nervous system. SNAP29 (synaptosomal-associated protein 29) is a t-SNAPE (soluble NSF attachment protein receptor) that is involved in intracellular transport in various vesicle and membrane fusion processes (Golgi apparatus, focal adhesions) (57).
F2; ARC syndrome. Arthrogryposis-renal dysfunction-cholestasis syndrome is clinically characterized by the association of arthrogryposis, renal dysfunction, cholestasis, and severe failure to thrive. Patients with this AR inherited multisystem disorder also develop severe ichthyosis in addition to a number of symptoms, such as deafness, platelet abnormalities, osteopenia, missing corpus callosum, recurrent infections, and dysmorphism. Most affected children die early. ARC syndrome is caused by mutations in the VPS33B (58) or VIPAS39 (59) genes, after which it is classified into ARCS1 and ARCS2. VPS33B and VIPAS39 play an important role in the biogenesis and function of lamellar bodies in the epidermis (60).
F3; Multiple sulphatase deficiency. Some metabolic diseases present, in addition to variable symptoms of different organ systems, also a more or less pronounced ichthyosis. The AR inherited multiple sulphatase deficiency is caused by mutations in the SUMF1 gene (sulphatase-modifying factor 1) and is one of the lysosomal storage diseases. The complex clinical picture develops usually only within the first 2 years of life, and in addition to a mild ichthyosis, includes symptoms such as metachromatic leukodystrophy and mucopolysaccharidosis. The diagnosis can be confirmed molecularly or biochemically by detecting increased excretion of mucopolysaccharides and sulphatides (61).
F4; Gaucher syndrome type 2. The presence of ichthyosis or a collodion membrane at birth has only been observed in the rare type 2 Gaucher syndrome (62). The further course of the disease is fatal, due to the occurrence of hepatosplenomegaly and progressive neurological symptoms, such as spasticity, seizures and oculomotor paralysis. Patients with Gaucher syndrome type 2 usually die before their second year of life. Genetic causes are AR inherited mutations in the GBA gene, which encodes the lysosomal enzyme beta-glucosidase (or beta-glucocerebrosidase), and plays a role in ceramide metabolism.
O1; KID syndrome. A rare autosomal dominant disease with keratitis, ichthyosis or hyperkeratosis and deafness. At birth, there is a collodion membrane or a non-ichthyosiform erythroderma. Characteristic lesions include progressive erythematous dermatitis with reddened, hyperkeratotic plaques, palmoplantar keratoderma, nail dystrophy, alopecia, and sparse or absent eyebrows and eyelashes. The typical verrucous aspect mainly affects the face, scalp, ears, elbows and knees. As genetic causes, mutations have been described both in the GJB2 gene (connexin 26) and in the GJB6 gene (connexin 30). These are mostly neo-mutations. Patients with KID syndrome appear to be at increased risk for squamous cell and tongue cancers. Mutations in the GJB2 gene may also result in other phenotypes, such as AR inherited deafness or AD inherited palmoplantar keratoderma type Vohwinkel (mutilating), depending on the location of the mutation (63).
O2; Chanarin Dorfman syndrome. Classified as a lipid storage disorder of neutral fats in which the breakdown of triglycerides in the cell is impaired (neutral lipid storage disease with ichthyosis; NLSDI). Due to the defect, lipid droplets accumulate in a wide variety of cell types; in granulocytes this characteristic phenomena is called Jordan’s anomalies. By demonstrating Jordan’s abnormalities in the blood smear, the diagnosis can be clinically made uncomplicated and cost-effective. The syndrome is inherited in an AR manner and is caused by mutations in the gene ABDH5 (CGI-58) (64). Patients with NLSDI are often born as collodion babies and later develop mild generalized ichthyosiform erythroderma (Fig. 2l) and hepatosplenomegaly. Symptoms such as hearing loss, cataract, nystagmus, mental retardation, and ataxia are less consistent. The accumulation of lipid vacuoles in skeletal muscle cells can lead to muscular complaints (muscle weakness, myopathy) with increasing age. Depending on the extent of liver and muscle involvement, the blood levels of liver and muscle enzymes are raised. In the differential diagnosis, if Jordan abnormalities are detected, another type of lipid storage disorder, similar to NLSDI, but with severe myopathy, and no ichthyosis (NLSDM) (65), should be considered. NLSDM is caused by mutations in ATGL (PNPLA2) (65).
O3; ARKID syndrome. The acronym ARKID syndrome stands for autosomal recessive keratoderma, ichthyosis and deafness. Some patients exhibit additional symptoms, such as mental retardation, microcephaly, short stature or hip dislocation. Genetic causes of ARKID syndrome are, as in ARC syndrome, mutations in the gene VPS33B (66), which is why ARC syndrome and ARKID syndrome are referred to as allelic diseases. All 4 patients described in the literature carried the same mutation at amino acid position 131 (p.Gly131Glu) on at least 1 allele (either homozygous or in combination with a different second mutation). Alter et al. published an 11-year-old patient with liver damage due to copper overload in addition to the well-known ARKID symptoms, as well as exocrine pancreatic insufficiency (67). Copper overload has not previously been reported with ARCID or ARC syndrome, but with MEDNIK syndrome.
O4; SAM syndrome. SAM syndrome is characterized by 3 predominant symptoms: severe dermatitis, multiple allergies and metabolic wasting. First, patients with biallelic loss-of-function mutations in the desmoglein 1 (DSG1) gene with an AR inheritance pattern were described (68, 69). As with Netherton syndrome, these patients have massively elevated levels of IgE. Later, cases of SAM syndrome were also diagnosed with heterozygous desmoplakin (DSP) gene mutations (70). Heterozygous mutations in DSP and DSG1 are known in AD transmitted striate palmoplantar keratoderma.
Congenital disorders of glycosylation (CDG) are due to deficiencies in the glycoprotein biosynthesis. The spectrum of clinical manifestation comprises multiple organ systems and includes ichthyosis. Four different types of CDG are caused by mutations in the genes MPDU1 (CDG-If), DOLK (CDG-Im), SRD5A3 (CDG-Iq) and PIGL, which is also known as CHIME syndrome or Zunich neuroectodermal syndrome (71, 72).
MPDU1-CDG is a defect in the N-glycan assembly in the endoplasmic reticulum (ER) (73, 74); patients present skin symptoms (ichthyosis, erythroderma), neurological features (psychomotor retardation, seizures), hypotonia, visual impairment, dwarfism and transient growth hormone deficiency.
DOLK-CDG is a defect in dolichol kinase that catalyse the last step of the dolichol phosphate biosynthesis. Patients have dilated cardiomyopathy, ichthyosis, epilepsy, microcephaly, visual impairment, hypoglycaemia and often die within the first 6 months (75, 76).
SRD5A3-CDG (cerebro-cerebello-oculo-cutaneous syndrome) is defined as a defect in polyprenol reductase within the biosynthesis of dolichol. Patients present ichthyosis, erythroderma and dry skin (77, 78).
Patients with PIGL-CDG or CHIME syndrome mainly have colobomas, congenital heart defects, early-onset migratory ichthyosiform dermatosis, mental retardation and ear anomalies. The defect is localized in the ER and concerns the second step of GPI-anchor biosynthesis, the de-N-acetylation of N-acetylglucosaminyl-phosphatidylinositol (79).
Inherited ichthyoses comprise a large spectrum of phenotypes that are caused by mutations in more than 50 different genes. Significant progress has been made in understanding the molecular mechanisms of ichthyoses over the past 25 years due to the accelerated development of DNA-based sequencing methods. However, there is still new evidence in genetics and molecular pathology of ichthyoses.
NGS has become a key technology for genetic testing and is applied in routine diagnostics of inherited diseases, since the costs are low, and the outcome is fast and effective. Multi-gene panel sequencing allows analysis of a few to a hundred genes simultaneously and cost-effectively, and guarantees the highest quality. The success rate of this method for the identification of disease-causing mutations in ichthyoses is approximately 80%. Large deletions or duplications cannot be fully detected by this method, although recent technical advances have been made to combine the evaluation of NGS and copy number variation (CNV) in the same analysis. Alternatively, CNVs can be detected by additional methods, such as multiplex ligation-dependent probe amplification (MLPA), quantitative real-time PCR, or aCGH (microarray-based comparative genomic hybridization). If multi-gene-panel analysis fails to detect disease-causing variants, WES can be used as an extended method, in which either only the DNA of the patient or, additionally, the DNA of the parents (trio) is examined. In WES all protein-coding regions (exons) of the approximately 22,000 human genes are analysed. The next possible level is WGS, which looks to the entire genetic information of the human genome, including complex structural variants. Structural variants comprise DNA segments inserted into or removed from the genome, as well as segments that are duplicated and segments whose direction is reversed. They are much more difficult to identify than single nucleotide variants, and it is actually not yet clear how many structural variants exist in a human genome.
The detection rate for disease-causing variants will increase with the help of all these performant technologies. However, the correct interpretation of the identified mutations is still associated with correct description of the phenotype and requires detailed clinical knowledge of diagnoses, differential diagnoses and terminology in dermatology.
The good news is that excellent clinicians will still be in great demand for the next 100 years.
The authors are grateful to Andreas Zimmer for technical support in the production of the figures.


 Click to show fullsize
Click to show fullsize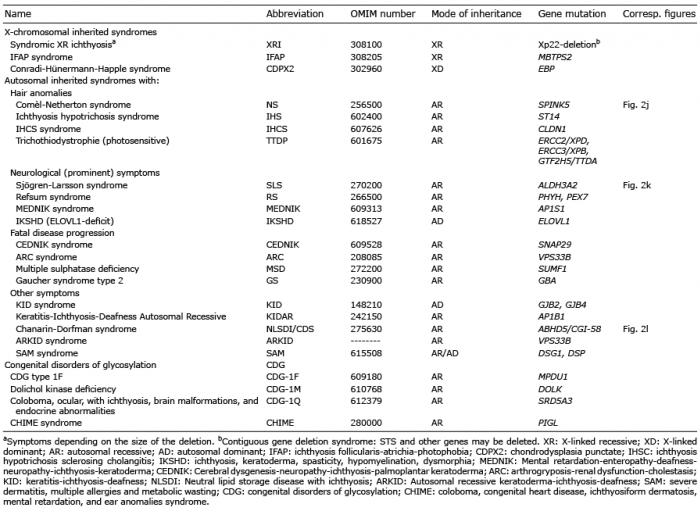 Click to show fullsize
Click to show fullsize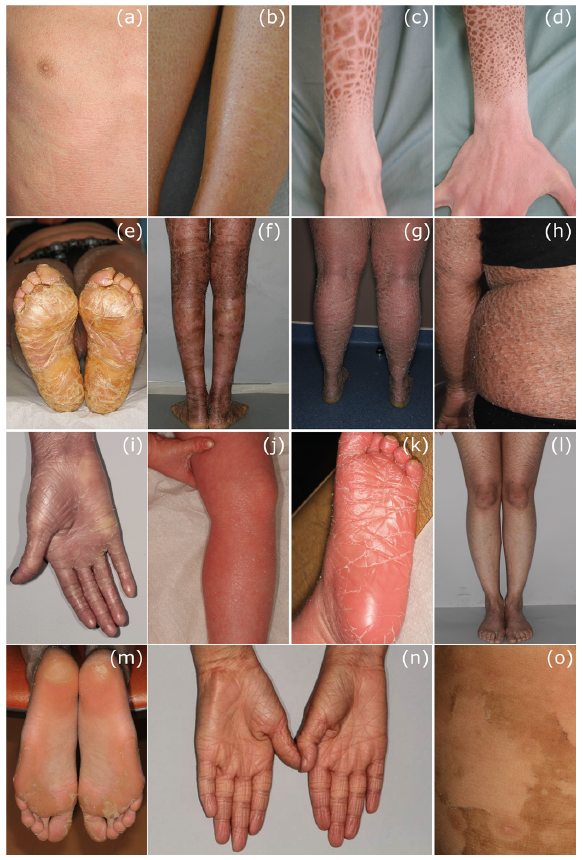 Click to show fullsize
Click to show fullsize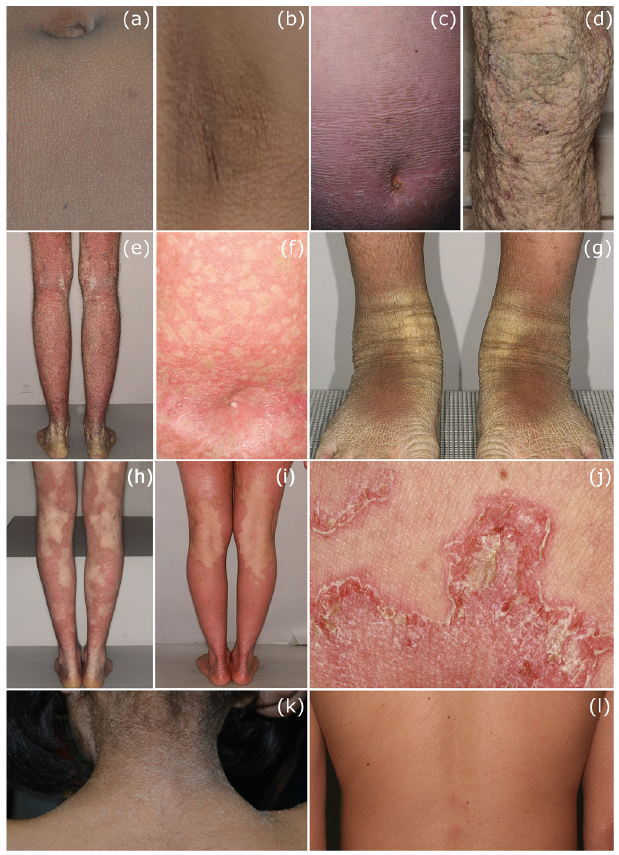 Click to show fullsize
Click to show fullsize