


Department of Medical Sciences, Uppsala University, Uppsala, Sweden
1This article is based partly on the Dohi Memorial Lecture presented at the 118th Annual Meeting of the Japanese Dermatological Association in Nagoya, June 2019.
The understanding of monogenetic disorders of cornification, including the group of diseases called ichthyoses, has expanded greatly in recent years. Studies of the aetiology of more than 50 types of ichthyosis have almost invariably uncovered errors in the biosynthesis of epidermal lipids or structural proteins essential for normal skin barrier function. The barrier abnormality per se may elicit epidermal inflammation, hyperproliferation and hyperkeratosis, potentially contributing to the patient’s skin symptoms. Despite this and other new knowledge about pathomechanisms, treatment of ichthyosis often remains unsatisfactory. This review highlights a series of approaches used to elucidate the pathobiology and clinical consequences of different types of ichthyosis, and related diseases with the ultimate goal of finding new and better treatments.
Key words: skin pH; ARCI; human epidermis; keratins; ceramides; therapy; epidermolytic; congenital; keratinocytes.
Accepted Feb 12, 2020; Epub ahead of print Mar 9, 2020
Acta Derm Venereol 2020; 100: adv00097.
Corr: Anders Vahlquist and Hans Törmä, Department of Medical Sciences, Uppsala University, SE-751 85 Uppsala, Sweden. E-mails: anders.vahlquist@medsci.uu.se, hans.torma@medsci.uu.se
Ichthyosis refers to skin diseases with scaling somewhat reminiscent of fish scales (Greek: ichthus=fish). There are more than 50 genetic types of, mostly non-syndromic, ichthyosis, ranging in severity and frequency from mild and common (prevalence < 1%) to severe and rare (< 0.001%). In the latter case, babies are often born with a thick horny layer (collodion), dermal inflammation and impaired skin barrier function, requiring intensive medical care. Nearly all patients with ichthyosis require daily applications of cream, sometimes complemented with retinoid tablets. This review highlights recent progress in the understanding of the causes and consequences of ichthyosis, which may lead to better care and treatments.
Ichthyosis is an umbrella term for more than 50 types of, usually monogenetic, diseases, all characterized by widespread hyperkeratosis, xerosis and scaling of the skin, sometimes also associated with syndromic features. Typically, the skin problems begin at birth or shortly thereafter and usually show lifelong persistence. Depending on the underlying genotype, disease intensity ranges from mild to severe, in the latter case markedly reducing the patients’ quality of life (1). Only rarely are there life-threatening consequences; for example, in neonates with harlequin ichthyosis (HI), epidermolytic ichthyosis (EI) and certain types of syndromic ichthyosis (2, 3). Later in life, less severe, but more common, complications occur, such as pruritus, ectropion and anhidrosis (Fig. S1). Careful medical attention is frequently required, including oral retinoid therapy. Yet, the vast majority of patients with ichthyosis have only mild to moderate skin symptoms, which are readily controlled by daily applications of cream (2, 3).
Despite a thick stratum corneum (SC), patients with ichthyosis usually have variably increased transepidermal water loss (TEWL). This is due to various defects in the biosynthesis of proteins and lipids essential for normal barrier formation, specific for each of the 4 main types of non-syndromic ichthyosis (4):
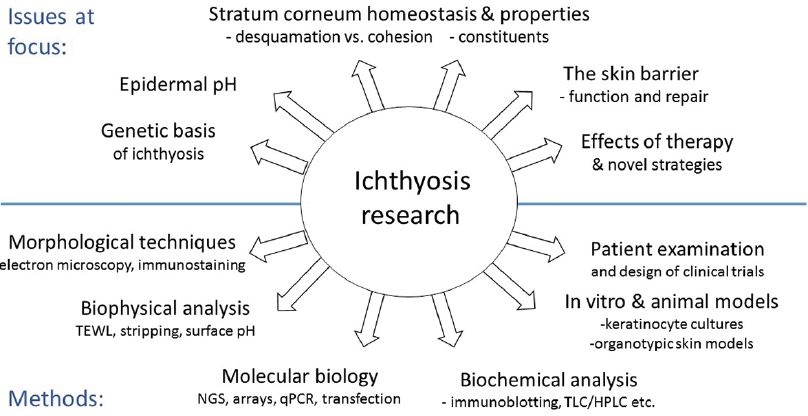
This overview exemplifies a wide range of approaches used to elucidate the aetiopathogenesis of various types of ichthyosis, research that concurently results in a better understanding of normal human skin biology and yields new ideas about dermatotherapy (Fig. 1).
Fig. 1. Examples of scientific issues and methodological approaches in ichthyosis research. NGS: next generation sequencing; LC: liquid chromatography.
The ingenious construction of human epidermis, with its many disparate functions and constant renewal of cells, also makes it vulnerable to genetic defects, frequently causing clearcut structural abnormalities. Thus, under light microscopy, I. vulgaris displays an absence of stratum granulosum (below SC) and in EI and ichthyosis with confetti due to various keratin mutations a clumping of intermediate filaments is seen, occasionally leading to cytolysis of suprabasal keratinocytes. However, in most other types of ichthyosis, electron microscopy (EM) is required to disclose the histopathological hallmarks (5–11).
In ARCI, for example, 4 distinctive ultrastructural patterns are identifiable in the granular and corneal layers of the epidermis: EM type 1 (lipid droplets), probably related to epidermal hyperproliferation (5); EM type 2 (“cholesterol clefts”(6)), typically associated with TGM1 mutations (12); EM type 3 (abnormal lamellar bodies and elongated membranes (7)), often associated with NIPAL4 mutations (9); and, EM type 4 (aggregated lipid membranes), exclusively associated with SCL27A4 mutations (13, 14). Furthermore, HI and related conditions due to ABCA12 mutations often show prominent distortions of the lamellar bodies (11, 15). Finally, and common to most types of ARCI, the lipid bilayers and CLEs are attenuated, best seen after ruthenium staining of the skin specimen (10).
EM analysis is clinically useful for differentiating ARCI from other conditions. For example, in a diagnostic team effort 2 Scandinavian half-brothers, initially believed to have an atypical form of ARCI, showed no signs of EM types 1–4, but the corneodesmosomes were few and abnormally looking (16). Genomic screening revealed novel mutations in the DSG1 gene, consistent with a mild form of SAM syndrome (severe dermatitis, allergy and metabolic wasting) caused by desmoglein deficiency (17). Another example concerns the rare disorder keratosis linearis, ichthyosis congenita with keratoderma (KLICK), which ultrastructurally exhibits massively enlarged keratohyalin granules (18). KLICK was eventually shown to be caused by recessive mutations in the regulatory elements of the POMP gene interfering with the proteasome degradation of numerous epidermal proteins (19).
Although EM is invaluable in many studies of ichthyosis, it is a tedious and costly method only available in certain laboratories. As an alternative, immunofluorescence (IF) analysis can be used, for example, for detecting cytoskeletal abnormalities in patients with ichthy-osis with confetti (20) or for experimental studies of cultured cells from patients with EI (21). Regarding the latter, Fig. S2 shows IF stainings of keratin 10 in differentiated keratinocytes from a patient with KRT10 mutation before and after in vitro exposure to heat. Clearly, heat stress causes aggregation of keratin filaments to a much higher extent than in healthy control cells (22). However, although the number of cellular aggregates was diminished by pre-treatment with a molecular chaperon designed to stabilize protein polymers (21), any extrapolation to the in vivo situation demands circumspection because the efficacy of topical chaperon was disappointing in a recent study of epidermolysis bullosa simplex, another keratinopathic disorder (23).
Invasive techniques are not always required for obtaining in vivo information about SC. By simply applying an evaporimeter and a flat glass electrode to the intact skin, measurement of TEWL, skin hydration (capacitance) and surface pH is possible. This low-tech approach is useful in both healthy and diseased skin; for instance, when studying the effects of various drugs and noxious agents potentially affecting the skin barrier (24, 25). Another finding from these studies is that TEWL is elevated in untreated ARCI skin and increases further after efficient treatment with topical keratolytics (26). While this might inadvertedly enhance the pathomechanism of ichthyosis, remaining amounts of SC seems nearly always to be sufficient for preventing any harmful losses of water or influx of toxic substances via the skin.
However, when SC is mechanically removed in toto down to the glistening layer of epidermis, TEWL will increase dramatically (27). For obvious reasons, a concurrent increase in pH from ~5 on the skin surface to 7.4 in viable epidermis must then also occur, although details of this event for long remained unexplored (28). Fig. S3 shows that, soon after a complete removal of SC, pH and TEWL will start to decrease again, reaching normal surface values within 5–7 days, approximately one week before the full restoration of SC (28). Indeed, pH appears to normalize more quickly than TEWL, possibly reflecting its master role during barrier repair. The importance of pH for SC homeostasis has also been highlighted by the discovery of a sigmoidal pH gradient over human SC, with its steepest slope occurring midway between stratum granulosum and the skin surface (28). This gradient, first demonstrated by repeated monitoring of pH in the course of >100 tape strippings, has since been confirmed using more sophisticated techniques in both human and mouse skin (29).
Interestingly, the pH gradient in SC looks quite different in I. vulgaris and XRI (30); in the former a shift towards less acidic values is observed, whereas the opposite is true for XRI (Fig. 2). The proposed explanation for this difference is a paucity of acidic break-down products of filaggrin (e.g. urocanic acid) in I. vulgaris and an accumulation of acidic CSO4 in XRI (30). Incidentally, CSO4 is a fascinating molecule, acting both as an inhibitor of SC desquamation (31) and as a signalling molecule during keratinocyte maturation (32, 33). In fact, a deficiency of CSO4 in epidermis due to recessive SULT2B1 mutations may also cause ichthyosis (34).
The key components contributing to the pH gradient in normal SC appear to be urocanic acid, free fatty acids and sodium-hydrogen exchanger -1 (NHE-1), all accumulating in acidic microdomains near the skin surface (35). Clearly, a reduction of approximately 2 pH units over a distance of only 10–20 µm (the normal thickness of SC) is biologically huge, and probably affects both lipid organization and protein structure at different depths of SC. Indeed, this makes treatment with pH-adjusting creams an intriguing option for some disorders of cornification (35–37). Examples of two pH-dependent enzymes operating in SC are kallikrein 5 and 7, the principal proteases involved in corneodesmosome degradation and desquamation (38). Besides pH, the activity of these enzymes depends on the amount of endogenous inhibitors, one of which is LEKTI (39). A genetic deficiency of LEKTI, as in Netherton syndrome (NS; Ichthyosis circumflexa), accelerates desquamation and reduces SC thickness to almost nil, hence dramatically increasing TEWL (39, 40). Ongoing clinical trials with topical application of synthetic inhibitors may lead to new treatments for NS and possibly atopic dermatitis, which is frequently associated with a secondary deficiency of LEKTI (41). Hypothetically, by modulating desquamation in the opposite direction, e.g. by blocking LEKTI, it might be possible to increase desquamation in some hyperkeratotic conditions, such as HI and EI, known to be associated with decreased secretion of proteolytic enzymes from the lamellar bodies (42, 43).
Fig. 2. The pH gradients over stratum corneum in healthy controls and patients with X-linked ichthyosis (XRI) or ichthyosis vulgaris (I. vulgaris). Mean pH values revealed by tape-stripping (n = 10–11 males in each group). When adjusting for the higher number of strippings required to remove SC in I. vulgaris (and XRI), the pH gradient will in the former case be shifted to the left compared to normal skin. (modified from refs. 28 and 30 with permission).
Considering the many different aetiologies of ichthyosis, it is not far-fetched to assume that homeostatic responses in epidermis will differ depending on the genotype and the extent of barrier insufficiency it causes. One way of testing this hypothesis is to study the global mRNA expression in epidermis, using microarray analysis of transcriptomes extracted from tissue biopsies and searching for differently expressed genes (DEGs) in ichthyosis compared with normal skin.
In such a recent study, microarrays consisting of 22,000 genes were applied to pooled skin extracts from healthy controls and untreated patients with either XRI or I. vulgaris due to mono- or bi-allelic FLG mutations (44, 45). While patients with XRI showed only 27 DEGs, patients with I. vulgaris showed up to 120 times as many DEGs (Fig. S4). Speculatively, the low number of DEGs in XRI is due to CSO4-induced hyperkeratosis reducing the need for more active barrier repair (46, 47). In I. vulgaris, since no “silent” generation of hyperkeratosis occurs, a chronic repair process takes place that might explain the abundance of DEGs. This hypothesis gains support from our gene ontology and qPCR analyses, showing activation of numerous genes involved in inflammation, lipid metabolism and hyperproliferation; the response is particularly evident in patients with biallelic FLG mutations who are also notoriously prone to develop eczema (24, 44).
When skin samples from patients with ARCI with TGM1 mutations were similarly investigated, a broad spectrum of 256 DEGs appeared; 25 involved in keratinization and cell mobility, 46 in immune response and 8 in acylCer biosynthesis, the last of which are also known as “ARCI genes” because of their involvement in the ARCI aetiology (48). Speculatively, a marked up-regulation of several ARCI genes reflects a positive feedback loop aimed at generating more omega-O-acylCer for barrier repair. However, in ARCI patients with truncating TGM1 mutations such a response is probably useless; no matter how many lipid precursors are available in the granular cells the absence of transglutaminase-1 (TGm-1) will prevent a proper crosslinking of CE and CLE (49).
Further support for a concept of ARCI proteins operating in a feedback regulated pathway comes from our recent studies using IF staining combined with CellProfiler imaging, allowing semi-quantitative comparisons of the protein expression at different depths of epidermis (48). Fig. 3 shows examples of results obtained in biopsies from 5 patients with TGM1 mutations and 4 healthy controls. Clearly 2 of the studied proteins, CYP4F22 and CerS3, co-localize in the granular layer of epidermis in both patients and controls, but the protein expression is much higher in the patients, thus corroborating the microarray data. The co-localization of 2 other ARCI proteins, TGm-1 and SDR9C7, was studied in more detail in healthy control skin using in situ proximity ligation assay (isPLA), which generates a signal when 2 different proteins are at a distance of less than 30 nm from each other (50). While filaggrin did not produce any isPLA signals with either of the 2 ARCI proteins, together they produced a strong signal in stratum granulosum consistent with a close interaction between TGm-1 and SDR9C7 in a chain of events leading to a proper formation of CLE (51). TGm-1 has also been found to co-localize with 12R-LOX and eLOX-3 in stratum granulosum of normal epidermis, but not in ARCI epidermis with inactivating mutations in NIPAL4 (encoding ichthyin) (52). This implies that ichthyin (a tentative transporter of Mg2+ (53)) is also essential for acylceramide synthesis, acting in close proximity to other ARCI proteins.
Fig. 3. Examples of immuno-fluorescence staining (left) and CellProfiler analysis (right) of the CYP4F22 and CERS3 proteins in patients with TGM1 mutations versus healthy controls. The increased expression in patients’ skin extends beyond the granular layer. The shaded area in the diagram correspond to stratum corneum (modified from ref. (48) with permission).
In addition to an increased expression of several wild-type ARCI genes, numerous other genes involved in barrier repair, lipid biosynthesis, inflammation and anti-microbial peptides (AMPs) defence are also heavily upregulated in ARCI epidermis (48, 54–56). Incidentally, increased expression of AMPs might explain why microbial infections are rare in patients with lamellar ichthyosis despite a fissured and scaly skin. Analogously, psoriatic lesions express high levels of AMPs, albeit in this case on a background of much stronger immune and inflammatory reactions (57).
However, not all subtypes of ARCI exhibit a resilience against bacterial infections. For example, patients with HI and IPS often experience neonatal skin infections and septicaemia; in this case possibly related to a defective release of AMPs from the lamellar bodies (43). Furthermore, skin infections are frequent in EI with intrinsic defects in barrier repair, inter-corneocyte lipid deposition and AMP release (43, 58, 59), although in this case skin erosions and blistering are certainly a major contributing factor.
By simply scraping the skin surface with a sharp blade, samples of SC can be collected for analysis of, for example, CSO4 (46), urocanic acid and natural moisturizing factors (NMFs) (60). Using slightly more invasive techniques, such as superficial shave biopsies, full-thickness samples of epidermis are obtainable without significant risk of scarring. After homogenization and extraction of such samples, sensitive analytical techniques, such as high-performance liquid chromatography (HPLC), allow quantitation of numerous endogenous compounds and drugs, such as vitamin A and retinoids. For example, reduced concentrations of retinol (vitamin A1) were found in I. vulgaris and increased levels of 3,4-didehydroretinol (vitamin A2) in some types of hyperproliferative keratosis (61, 62), as yet without known significance. Although endogenous concentrations of all-trans retinoic acid in epidermis usually fall below the detection limit of the assay, therapeutic levels of isotretinoin and acitretin can be measured in shave biopsies (63, 64).
Using more sophisticated detection methods, such as ultra-performance liquid chromatography and mass-spectrum detection, the skin levels of fatty acids of different chain lengths, squalene and various types of ceramides (Cer) are quantifiable with high sensitivity and specificity (65–67). Indeed, the abnormal levels of various ceramides found in ARCI epidermis gave early clues to the existence of inborn errors of acylCer biosynthesis (68), which was later confirmed via gene hunting. Fig. 4 summarizes our current understanding of lipid barrier formation in epidermis and the critical positioning of several ARCI proteins (for review see (4, 69, 70)). Subsequent to the enzymatic elongation of fatty acids (FA) by ELOVL4, the ultra-long chains (ULCFA) form amid-linkages with sphingosines, hence constituting Cer. This highly hydrophobic molecule undergoes a series of modifications, including a CYP4F22-mediated ω-hydroxylation of the FA moiety and subsequent transacylation with linoleic acid to form acylCer (71). The latter step, enhanced by PNPLA1, is essential for the formation of the lipid bilayers in SC. A significant fraction of acylCer undergoes further oxidation of the linoleate moiety, catalyzed by 12R-LOX and eLOX-3, and a subsequent covalent binding to CE (72). This final step in CLE formation was previously thought to be catalysed by TGm-1, analogous to the transacylation of involucrin. However, a recent report implicates an alternative pathway involving SDR9C7 (67). SDR9C7 is a dehydrogenase, that converts the oxidated linoleate molecule into a 13-ketone, a reactive moiety known for its non-enzymatic coupling to protein (67). As a corollary, ARCI caused by SDR9C7 deficiency is characterized by absent CLEs on EM examination (67).
Fig. 4. Crucial components and biosynthetic steps in the formation of an epidermal lipid barrier. Light green (hatched) arrows in the box indicate two alternative pathways in cornified lipid envelope (CLE) formation (modified from Am J Clin Derm (4) with permission). SC: stratum corneum, SG: stratum granulosum, ECM: extracellular matrix; CE: cornified envelope, FFA: free fatty acids, ULC-FA: ultra-long chain fatty acid, AcylCer: acyl ceramide, ELOVL4: ELOVL fatty acid elongase 4, CYP4F22: cytochrome P450 family 4 subfamily F member 22, FATP4: fatty acid transport protein 4, CERS3: ceramide synthase 3, PNPLA1: patatin-like phospholipase domain containing 1, 12R-LOX: arachidonate 12-lipoxygenase, 12R-type, eLOX-3: hydroperoxide isomerase ALOXE3, TGm-1: transglutaminase-1, NIPAL4: magnesium transporter NIPA4 (ichthyin), LIPN: lipase member N, SDR9C7: short-chain dehydrogenase/reductase family 9C member 7.
Interestingly, Crumrine et al. (70) recently proposed that virtually all the above-mentioned processes take place within the lamellar bodies, subsequently delivering preformed CLE scaffolds and lipid bilayers to the intercellular space via exocytosis. It was also suggested that some previously unexplained ultrastructural features in ARCI are actually caused by toxic levels of free FA accumulating in the keratinocytes owing to a downstream blockade in the acylCer pathway (70). As a possible extension to this “blockage theory”, our own findings of an upregulation of several ARCI proteins in the skin of patients with inactivating TGM1 mutations (48) imply that lipoxygenated acylCer, instead of being converted to CLE by TGm-1, accumulates in the corneocytes as lipid aggregates or membranes. Speculatively, this might explain some of the EM characteristics of TGM1-associated ARCI (6). Conversely, more upstream blockages of acylCer biosynthesis, e.g. due to CYP4F22 or CERS3 mutations, might instead reduce the acylCer levels and thus impair the formation of both intercellular lipid bilayers and CLE. Although much remains to be clarified about this and the other pathogenic process in ARCI, there are already good arguments for distinguishing aetiologies related to inborn errors of the acylCer metabolism from other causes of ARCI; for example, by using prefixes, such as “lipodysgenic” or “lipid synthetic”, for this groups of disorders (48, 70).
Thanks to research mainly from France, Germany, Japan, Scandinavia, UK and the USA, it is now possible to genetically diagnose all forms of common and keratinopathic ichthyosis, and 85–90% of cases with ARCI (for review see (4)). With respect to the latter diagnosis, the leading causes of ARCI in Northern Europe are homozygous or compound heterozygous mutations in TGM1 (30–35%), ALOX12B or ALOXE3 (combined 15–20%) and NIPAL4 (12–15%) (4, 73–76). Detailed discussions about the genetics of ichthyosis are available in 2 other papers (77, 78).
Patients with ichthyosis frequently exhibit skin signs and symptoms that may be difficult for the examining doctor to describe in a concise way or to score for severity grade, e.g. in relation to a scientific study. Characteristically, skin lesions may be generalized or only occur focally, and the intensity of scaling may range from mild to severe, with scales typically described as lamellar, collodion-like, cobblestone-like, brownish, fine and white, etc. Furthermore, a plethora of other symptoms occurs, such as xerosis, palmoplantar keratoderma, erythema, itch and pain, all with a variable degree of severity. Adding to this complexity, phenotypic fluctuations often occur over time, either spontaneously or as the result of treatment or environmental factors, e.g. work, climate and season of the year. No wonder then a consensus is still lacking about the best severity scoring system to use in clinical trials (for review see (79))
In clinical practice, however, less sophisticated scoring models may still be useful. For example, in a recent study of 132 patients with ARCI, separate scorings (0–4) of ichthyosis (IS) and erythema (ES) severity were made in 10 different body regions, followed by an area-adjusted summation of individual score values (4, 73, 80). When the IS and ES values recorded at age >1 year were plotted against one another in a diagram, the individual ratios roughly distributed into 4 partially overlapping circles seemingly corresponding to the major clinical subtypes identified at first examination, i.e. before the genetic results became known (Fig. 5). Unsurprisingly, harlequin ichthyosis (HI), the rarest and most severe subtype of ARCI due to truncating ABCA12 mutations, shows the highest IS and ES values. Lamellar (LI) and erythrodermic ichthyosis (CIE), with more varied and partially overlapping phenotypes and genotypes, show high values of either IS or ES. The 4th entity, shows low values of both IS and ES, although most of the patients had severe skin symptoms at birth, healing spontaneously over a period of several weeks. This altering pattern is consistent with pleomorphism, “a condition in which an individual assumes a number of different forms during its life-cycle”. Accordingly, pleomorphic ichthyosis (PI) is a suggested new name for this subgroup of ARCI previously known as “non-LI/non-CIE” (80). It comprises several distinct conditions, such as self-improving collodion ichthyosis (mostly due to mild ALOX12B mutations), bathing-suit ichthyosis (due to temperature-sensitive TGM1 mutations) and IPS (specifically caused by SLC27A4 mutations) (73, 80, 81). (Nb: “syndrome” is probably a misnomer for IPS, because all extra-cutaneous symptoms appear to be secondary to the skin malfunction.)
While a crude classification of ARCI into 4 major subgroups may seem superfluous in an era of exact genetic diagnosing, it is still useful; for instance, when diagnosing ARCI without available genetic expertise or for teaching medical students how to distinguish between the various types of ichthyosis.
Fig. 5. Tentative correlation between ichthyosis and erythema severity 4 types of autosomal recessive congenital ichthyosis (ARCI) with partially overlapping phenotypes and culprit genes. (modified from refs. (4, 75) with permission).
Another bonus of a detailed skin examination is the chance of making serendipitous findings. Fig. 6A illustrates such a case: a 45-year-old woman, diagnosed in childhood with keratitis, ichthyosis, deafness (KID) syndrome due to a recurrent mutation in GJB2 (82). She started in her 20s to develop spots of normal-looking skin, which gradually grew in size and number, were histopathologically “non-lesional”. A subsequent sequencing of DNA from the healed spots revealed several de novo mutations restricted to the disease-causing GJB2 allele. Co-transfection of germline and de novo (somatic) mutations together with wt-GJB2 in HeLa cells showed that the de novo gene product remained intracellular, thus allowing an unopposed incorporation of wild-type connexin 26 into the gap-junctions (Fig. 6B) (83). Similar examples of spontaneous revertants in the skin have been described in epidermolysis bullosa (84), ichthyosis with confetti (85) and loricrin keratoderma (86). This makes drug enhancement of revertance (“natural gene therapy”) an interesting possibility for dominant negative genodermatoses (87, 88).
Fig. 6. Patient with keratitis-ichthyosis-deafness (KID) syndrome showing spontaneous revertant GJB2 mutation restoring the normal phenotype of keratinocytes. (A) Gradually appearing white spots in erythrokeratodermic areas on the thigh, and (B) effects of the patient’s somatic (silencing) mutation on an allele with germline GJB2 mutation (EGFP) when transfected to HeLa cells together with wt-GJB2 (Cherry). The blurred gap junctions (top panel), resembling the situation in lesional skin, are restored by the de novo somatic mutation (bottom panel) as in healed spots (modified from ref. (83) with permission).
Besides emollients and keratolytic creams (2, 3, 26), retinoids remain mainstay therapy for moderate to severe forms of ichthyosis. Acitretin and isotretinoin are the preferred drugs for systemic use, with newcomers, such as alitretinoin, probably having a less favourable risk/benefit ratio (89), and retinoic acid metabolism blocking agents (RAMBAs), such as liarozole, not yet commercially available (90). Broadly speaking, vitamin A agonists have anti-keratinizing and keratolytic effects. However, because many retinoids bind to specific ligand-activated transcription factors and regulate the expression of numerous genes expressed in epidermis, more specific effects on ichthyosis pathogenesis are also to be expected. One example is the different outcome of retinoid treatment in patients with epidermolytic ichthyosis due to KRT10 or KRT1 mutations (91). Whereas the former patients respond quite well to low-dose retinoid therapy, consistent with a down-regulation of mutated KRT10 (92), patients with KRT1 mutations often get worse and develop more blisters during retinoid therapy (91). A proposed explanation is the ubiquitous down-regulation of KRT2 by retinoids; this effect is harmless in both normal and KRT10-mutated epidermis, but deleterious in patients with KRT1 mutations who depend on keratin 2 as a replacer of mutated keratin 1 during its dimerization with keratin 10 (93). Conversely, patients with the Siemens type of superficial ichthyosis, caused by keratin 2 mutations that interfere with its heterodimerization to keratin 9, are known to respond most favourably to retinoids (94).
Encouragingly, many new ideas for ichthyosis treatment are in the pipeline, targeting not only the causative mechanisms, but also secondary events, such as inflammation and hyperproliferation. Although gene therapy for skin diseases has not yet proved as successful as initially hoped, topical antisense therapy blocking the translation of mutated mRNA has shown promising results, at least in pachyonychia congenita, a keratinopathic disorder mechanistically similar to epidermolytic ichthyosis (EI) (95). Moreover, disruption of mutated KRT10 in EI keratinocytes using a transcription activator-like effector nuclease (TALEN) technology reverts the intermediate filament fragility in vitro (96). These and other approaches, such as CRISPR/Cas9 gene editing, aimed at correcting the underlying mutation in situ, holds promise for a more specific gene therapy for ichthyosis in the future (97, 98).
Substitution and replacement therapy are other interesting approaches. Since various types of ceramides can now be synthetized in large amounts, they are obvious candidates for testing topically in ARCI (99). Another, still preclinical, approach is to enhance the acylCer pathway via ligand stimulation of transcription factors, such as peroxisome proliferator activating receptors (PPARs) expressed in epidermis and known to affect the expression of many ARCI genes in vitro (100). Several PPAR agonists are already in use for diabetes and cardiovascular disease. However, for this hypothetical treatment to be effective in ARCI, all genes involved in CLE formation must remain at least marginally intact, implying that lipodysgenic ARCI due to truncating mutations will remain unresponsive. In this context, enzyme replacement therapy (ERT) with topically applied recombinant transglutaminase may become an attractive (but expensive) future option, especially for patients with TGM1-associated ARCI (101). Perhaps a combination of ERT and supplementation with synthetic ceramides would prove most versatile, although this approach remains to be studied.
As regards treatment of secondary pathogenic events, it is noteworthy that skin inflammation in ARCI has many similarities to psoriasis, making already approved biological therapies feasible to test in severe cases of ichthyosis (56). In the long term, the search for new therapies in ichthyosis should also focus on alternative ways to restore the skin barrier and to dampen excessive intrinsic responses, which often cause more harm than relief to the patient. Whether this goal is attainable through gene technology and new biologics, or by specifically tailored molecules and substitution therapies, remains to be determined. Fig. 7 gives a summary of these and other prospects for ichthyosis treatment.
Fig. 7. Summary of ideas about future development of ichthyosis therapy. ERT: enzyme replacement therapy.
The skin is, both clinically and pre-clinically, a “research-friendly” organ. By combining a wide variety of investigative methods, ranging in complexity from simple in vivo measurements of TEWL and surface pH, to high-tech biochemical and genomic analyses of minimally invasive skin biopsies, or in vitro cultures of reconstituted skin, much information is attainable about the pathobiology of many skin diseases, not least ichthyosis.
Today, when almost 100 subtypes of ichthyosis have been characterized at both the genomic and ultrastructural level, an exact diagnosis early in life, a definite establishment of mode of inheritance, and an accurate genetic counselling should nearly always be feasible.
Though the treatment options have also evolved over the years, there is still a great need for new developments aimed at improving the patients’ quality of life. Through this research, new knowledge may also be gained about many other skin diseases with biological features similar to ichthyosis, such as eczema and psoriasis, which are also characterized by inflammation and a perturbed skin barrier.
We thank our co-authors and the participating patients, health & laboratory staff, and funders for all their support in the presented studies.
Conflicts of interest: AV is the Chairman of the Society for Publication of Acta Dermato-Venereologica.


 Click to show fullsize
Click to show fullsize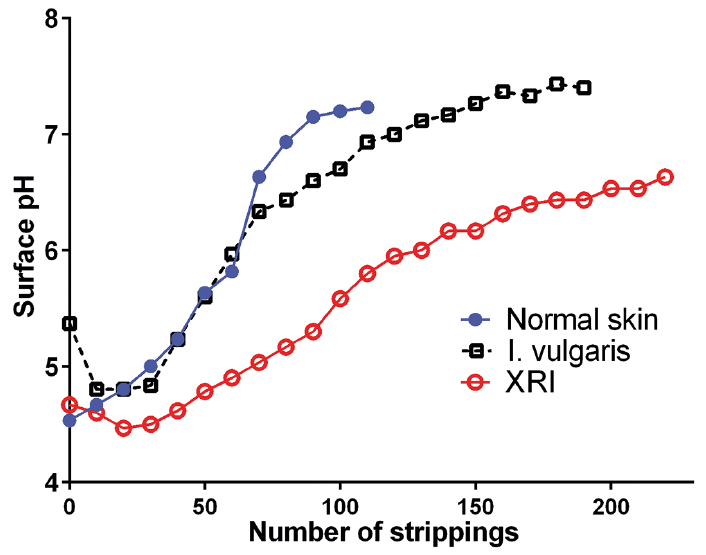 Click to show fullsize
Click to show fullsize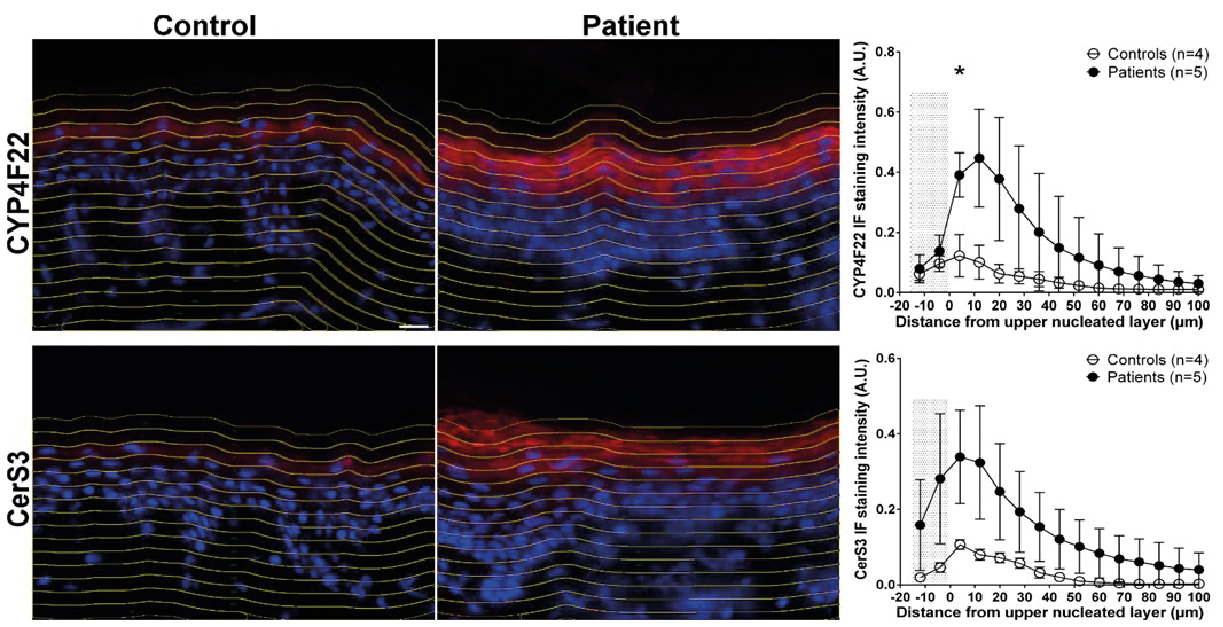 Click to show fullsize
Click to show fullsize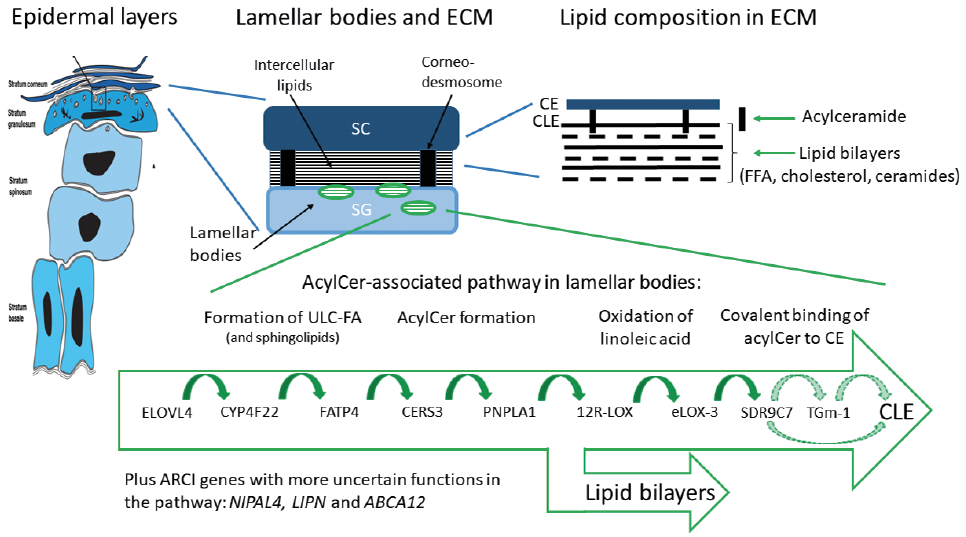 Click to show fullsize
Click to show fullsize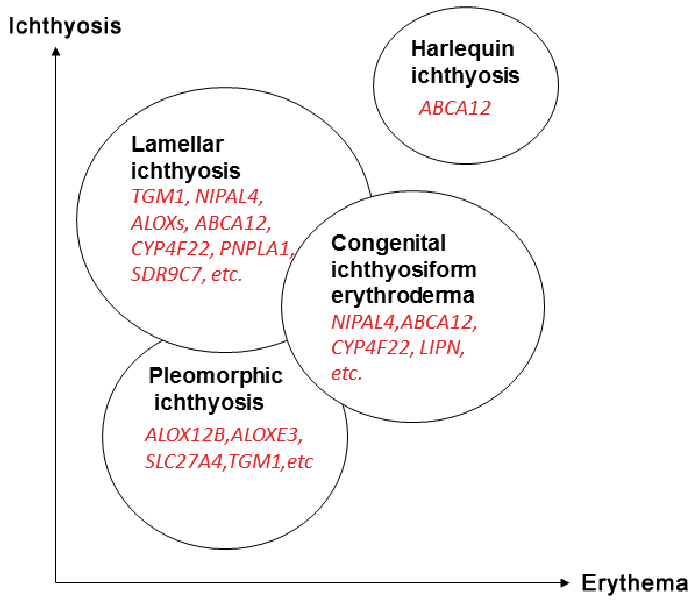 Click to show fullsize
Click to show fullsize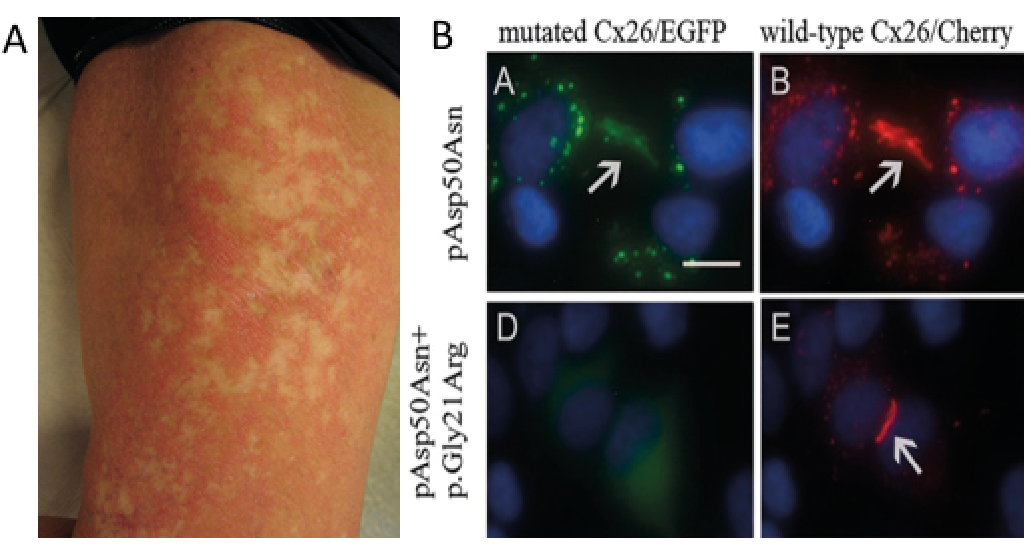 Click to show fullsize
Click to show fullsize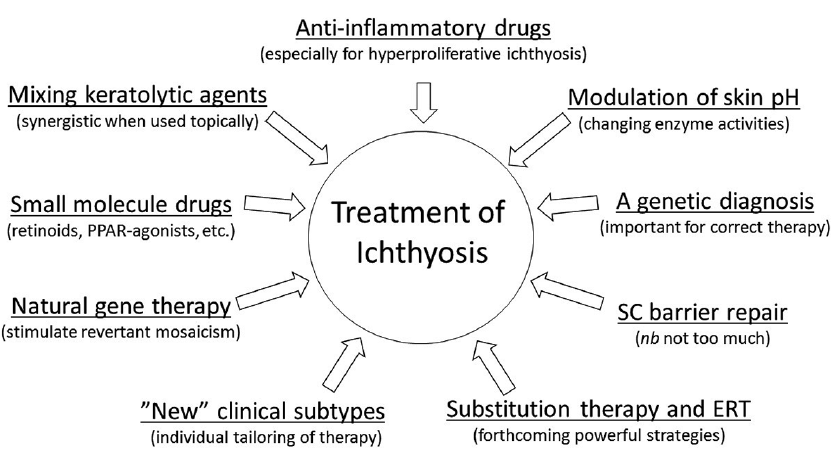 Click to show fullsize
Click to show fullsize