


Cambridge Cancer Centre, Cambridge University Hospitals NHS Foundation Trust, Cambridge CB2 0QQ, UK
Introduction of new systemic therapies in the last 10 years has radically improved outcomes for melanoma patients. Even so, not all patients benefit, so getting the right treatment to the right patient is a priority. These two major drug classes, small molecule targeted kinase inhibitors and immune checkpoint inhibitors, both come at significant cost, with sometimes serious side effects as well as high expense for health services. Almost half of melanomas harbour a BRAFV600 mutation and virtually all patients receiving BRAF targeted therapy will experience some amount of response. However, duration of response with these agents is uncertain, due to acquired resistance, which means few patients remain in response long term. Most metastatic melanoma patients are potentially eligible for immune checkpoint inhibitors, irrespective of BRAF status. However, only about half of patients will respond to these agents, and only half again will benefit long term. Thus, both primary and acquired resistance limit response. In this era of personalized anti-cancer therapy, biomarkers offer a means to predict for both response and relapse to a particular treatment. To date, the only validated biomarker applied to selecting melanoma systemic therapy is the BRAF gene. However, modern technologies are now opening up a wide range of candidate genes, polypeptides and proteins which are being evaluated for their potential clinical application as predictive biomarkers of the future.
Key words: melanoma; biomarkers; immunotherapy; BRAF targeted therapy; response.
Accepted Apr 27, 2020; Epub ahead of print Apr 28, 2020
Acta Derm Venereol 2020; 100: adv00142.
Corr: Dr Pippa Corrie, Cambridge Cancer Centre, Cambridge University Hospitals NHS Foundation Trust, Cambridge CB2 0QQ UK. E-mail: pippa.corrie@addenbrookes.nhs.uk
Systemic therapy options for melanoma patients are rapidly increasing. They offer life extension for many, but not all patients benefit. These high cost drugs also have complex, life-changing and potentially life-threatening side effects. Modern ‘Precision Medicine’ aims to personalize therapy for individuals and hence offer the opportunity to selectively treat only those expected to benefit from a particular therapy, while avoiding exposure to ineffective treatment in others. To date, the only validated predictive melanoma biomarker guiding treatment decisions is the BRAF gene mutation, although emerging modern technologies are identifying many more candidates whose clinical application have yet to be ascertained.
In the last decade, treatment of metastatic melanoma has undergone unprecedented transformation, with two new classes of anticancer drugs entering routine clinical practice, tripling overall survival of people whose life expectancy previously was limited to under one year. Both sets of drugs – BRAF targeted therapies and immune checkpoint inhibitors – are now being offered earlier in the disease pathway, to people who have undergone surgery for locoregional melanoma, based on evidence that adjuvant therapy halves the rate of recurrence (1–3). Despite this positive outlook, there are serious limitations yet to be overcome: little more than half of metastatic melanoma patients embarking on systemic therapy will achieve durable response, drug-induced toxicity can be life-threatening and certainly life-changing, while the cost of chronic drug prescribing is crippling many healthcare systems.
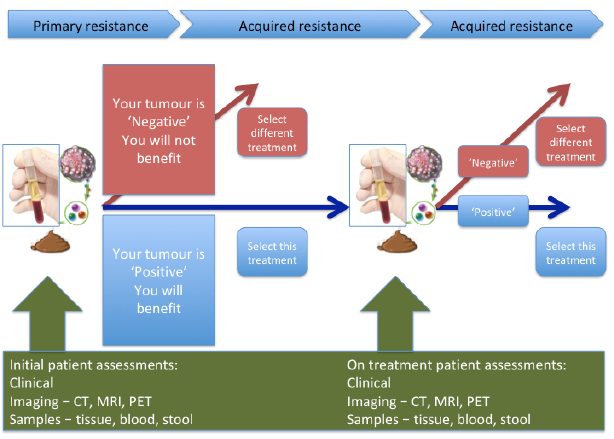
This same decade has seen a massive step change in our understanding of cancer biology. We are now in the era of ‘Precision medicine’, which aims to personalize treatment based on specific biological characteristics of an individual and their cancer. So-called biomarkers should, in theory, enable preferential selection of effective treatment, while avoiding exposure to inactive drugs causing unnecessary side-effects, thus also contributing to more cost-effective healthcare. Primary and acquired resistance to both molecularly targeted agents and immunotherapy limit treatment response. Therefore, biomarkers may be valuable adjuncts to clinical decision-making both prior to initiation of treatment, as well as during treatment, to predict the likelihood of treatment failure and disease relapse (Fig. 1). In practice, despite an explosion of research in this field, the role of predictive biomarkers in the clinic currently remains limited. The case of modern melanoma therapeutics well illustrates both the successes and challenges of biomarker discovery and their application.
Fig. 1. Integrating biomarkers into routine clinical practice.
In the whole of modern drug development, the mutant BRAF gene stands out as a massive success story in biomarker discovery. In 2002, a team at the Wellcome Sanger Institute reported BRAF mutations in 66% of melanoma cell lines tested and these findings were subsequently corroborated in melanoma patients (4). Its success as a treatment response biomarker is thanks to a talented biochemist who designed a drug to specifically block the active kinase domain of the mutant BRAF protein. This ‘lock and key’ approach generated ground-breaking responses in BRAF mutant metastatic melanoma patients treated in the phase 1 trial of the first specific BRAF kinase inhibitor, vemurafenib (5). In subsequent large-scale randomised trials, BRAF-targeted kinase inhibitors have generated objective response rates of up to 70% with virtually all treated patients experiencing some degree of response (6). The limitation of BRAF inhibition, however, is duration of response, due to onset of secondary resistance in most cases within a year of starting treatment.
Molecular characterization of tumours biopsied at the time of disease progression showed that reactivation of MEK downstream of BRAF was a consistent feature. Dual blockade with BRAF and MEK inhibitor combination regimens delay onset of secondary resistance, significantly extending duration of response (6). Unequivocal evidence that mutant BRAF drives malignancy in some 45% of melanomas led rapidly to adoption of BRAF testing of patient’s tumour tissue into routine clinical practice worldwide. Progression biopsies identified emergence of new mutations associated with loss of treatment response, some of which might be actionable and offer options for subsequent treatment.
However, accessing tumour is not always practical and is fraught with issues, particularly around tumour heterogeneity. Measuring circulating tumour DNA (ctDNA) in plasma as a ‘liquid’ biopsy offers an attractive, less-invasive alternative surrogate for disease burden. Preliminary studies support mutant BRAF ctDNA as a biomarker predicting for minimal residual disease and recurrence after surgical resection of locoregional melanoma (7) as well as lending value to monitor metastatic melanoma patients on treatment (6, 8), for early signs of both response and disease progression. Although a significant step change in patient management, work is still needed to optimize and standardize liquid biopsy methodologies, while larger scale prospective trials are essential to fully determine the clinical application of ctDNA before being introduced into routine clinical practice.
Other less common driver mutations occurring in melanoma include NRAS, PTEN loss and CKIT. Despite attempts to block signalling from these aberrant pathways, clinical benefits have been modest and no targeted agents have yet been approved for patients with these molecular characteristics. Currently, therefore, their significance as biomarkers is confined to research studies.
In contrast to molecular targeted agents, and also to some other cancers for whom they are approved, access to immune checkpoint inhibitors is not limited by any biomarker-determined subgroup of melanoma patients. Since first tested in melanoma patient trials, eligibility has been primarily determined by concerns for patient safety, as well as enrichment for better prognostic groups. Outside of clinical trials, real world experience has widened access and together with increasing understanding of how checkpoint inhibitors work, some clinical features have emerged that may help predict for benefit. This is particularly pertinent for BRAF mutant melanoma patients, who must choose which order to access the two drug classes available to them.
Immune checkpoint inhibitors rely on activating cytotoxic (CD8+) T-cell function, which can take a few weeks to kick in after initiating therapy. Evidence suggests that patients with slowly progressing, low disease burden (reflected in routine clinical and laboratory parameters including good performance status, normal serum lactate dehydrogenase, few organs involved, non-visceral disease) tend to respond to checkpoint inhibitors better than patients with high burden, rapidly progressing disease. These factors are readily identifiable in the clinic, but mainly reflect overall disease prognosis. Similarly, they predict for better outcomes with BRAF-targeted therapy (9) (Fig. 2). A recent meta-analysis of advanced melanoma interventional registration trials of systemic targeted therapies and checkpoint inhibitors demonstrated that BRAF-targeted therapies offer superior overall survival in the short term, which may be the priority for those patients with more aggressive disease and poorer prognosis, but checkpoint inhibition offers longer term survival gains for those who respond (10). However, given complex toxicities, high drug cost and limited overall survival benefits, there is a pressing need to utilise modern scientific capability to select the right treatment for the right patient based on their individual disease biology.
Fig. 2. Impact of tumour burden (as defined by lactate dehydrogenase (LDH) and number of body organ sites affected) on overall survival (OS) following treatment with dabrafenib+trametinib. ULN: upper limit of normal. (Reprinted with permission from The New England Journal of Medicine, Caroline Robert et al., Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma, 381:626-636. Copyright © (2019) Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society).
Increasing numbers of melanoma patients are receiving immune checkpoint inhibitors as their first line of treatment both in the adjuvant and advanced setting, striving for long term survival benefits. The dominant agents in clinical use are the anti-PD-1 antibodies, nivolumab and pembrolizumab (6). Both are generally well tolerated in all age groups, so in this modern age, advancing years is not a barrier to access and the numbers of melanoma patients being treated worldwide is rising exponentially, despite relatively modest benefits: response rate in metastatic melanoma is around 40%, while only the minority of those patients receiving adjuvant anti-PD-1 monotherapy are likely to benefit (1,2). Identifying the subgroup of patients expected to respond is a major research priority. Anti-PD-1 agents are licensed to be administered until disease progression, but chronic drug administration is driven by Pharma, not by biology. Can biomarkers also help determine treatment duration for an individual patient?
As a strategy to enhance activity, nivolumab (nivo) was combined with the anti-CTLA-4 antibody, ipilimumab (ipi) and the combination (ipi+nivo) regimen was compared to both monotherapies in the CheckMate 067 international registration trial. Response rates with the combination regimen were higher, reaching 58% for ipi+nivo compared with 45% for nivo and 19% for ipi, but the overall survival gain with ipi+nivo compared with nivo alone was marginal: 4-year overall survival 53% versus 46% (11). On the other hand, ipi+nivo was associated with a three-fold increase (59% versus 22%) in severe or life-threatening adverse events, compared to nivo alone, while 40% and 12% of patients discontinued treatment due to adverse events in these 2 trial arms. There is therefore a pressing need to identify those patients unlikely to benefit from the combination regimen to avoid unnecessary treatment-related toxicity.
In the last 5 years, a huge amount of resource has been invested in better understanding tumour immunology with significant focus on identification of biomarkers to address the questions posed here. As summarised by Chen & Mellman (12), cancers can be categorized into 3 groups: 1) ‘hot’ or inflamed tumours, characterized by a high T-cell infiltrate, 2) ‘cold’ or non-inflamed tumours, devoid of any T-cell infiltrate, and 3) cancers that have T cells and other immune cells present, but only at the periphery or within the stromal tissue and not within the tumour itself (Fig. 3). ‘Hot’ tumours are most likely to respond to checkpoint blockade, and melanomas fall in to this category. However, overall, the minority of melanoma patients respond to checkpoint blockade, demonstrating that the relationship between the tumour, host and microenvironment is hugely complex and no perfect biomarker of response or toxicity is yet available for clinical application. Highlights of expansive research in biomarker identification have been reviewed in various recent publications (for example, see 13–15). While not meant to be an exhaustive list, the role of the most promising biomarkers is summarized here (Table I) under these 3 headings.
Fig. 3. The tumour immune-microenvironment can be classified as being either (A) immune-excluded, (B) inflamed, or (C) non-inflamed. (Reprinted with permission from Springer Nature: Nature Medicine (Understanding the tumor immune microenvironment (TIME) for effective therapy, Mikhail Binnewies et al. (63), COPYRIGHT (2018)).
Table I. Summary of potential melanoma predictive biomarkers
Programmed death ligand 1 expression
Programmed death ligand 1 (PD-L1) is a protein expressed on cancer cells, tumour infiltrating lymphocytes (TILs) and myeloid cells which, through engagement with its receptor, PD-1, attenuates T-cell responses, thereby helping cancer cells evade immune surveillance. Anti-PD-1 antibodies disrupt PD-1:PD-L1 interactions to reinvigorate T-cell cytotoxicity. PD-L1 expression was therefore the first tumour-associated protein to be explored as a putative biomarker of response to anti-PD-1 antibodies. Initial analysis in the CheckMate 067 trial suggested that patients with high levels of PD-L1 had higher response rates compared with those whose tumours had low, or no expression (16). However, responses still occurred among these patients with low/no expression and the predictive value of PD-L1 expression was not borne out with longer follow-up (11). Since CheckMate 067 was initiated, the limitations of PD-L1 testing have received much attention: which antibody, which cells to count (tumour, immune cells, or both), which cut-off to use (cell count is linear, not binary) and all lack clarity. While in some other cancers PD-L1 expression does appear predictive, currently there is no place for routine testing in melanoma clinical practice.
Tumour mutational burden
Response to immune checkpoint inhibitors is highest among tumour types with a high mutation load and melanomas generally have high levels of mutations (17). This may be attributable, at least in part, to the production of tumour-specific neoantigens. Mutations within a tumour may lead to the formation of peptides unique to tumour cells that have the potential to be antigenic. Therefore, an increase in the tumour mutational burden (TMB) of a tumour could increase the likelihood of production of antigenic tumour-specific peptides, in turn leading to a larger pool of tumour-specific T cells. This larger pool of tumour-specific T cells would theoretically produce a greater antitumor response on inhibition of immune checkpoints that may be mediating tumour immune tolerance.
The first confirmatory human data came from whole-exome sequencing of DNA from tumours and matching blood from 25 metastatic melanoma patients treated with ipilimumab (18). There was a significant difference in TMB between patients with a long-term clinical benefit and those with minimal or no benefit, which was then reproduced in a subsequent validation set. High TMB was subsequently shown to correlate with survival following anti-PD1 blockade (19). Even so, as with PD-L1, measuring TMB is not straightforward. Gene sequencing methodology – which platform to use, which cut-off for a non-binary measure – is still evolving. Tumour heterogeneity will influence any measure of TMB in a discrete tumour sample, although some early research suggests this could be overcome by measuring TMB in a blood sample. Therefore, TMB remains an exploratory biomarker for the time being.
Aberrant signaling pathways driven by tumour mutations
Genetic mutations within melanoma cells have downstream effects on signalling pathways, which influence response to immunotherapy. A key pathway implicated in resistance to both anti-PD-1 and anti-CTLA-4 antibodies is the WNT/β-catenin-signalling pathway (20) which induces T-cell exclusion. Studies have demonstrated that loss of PTEN correlates with decreased T-cell infiltration at tumour sites, reduced likelihood of successful T-cell expansion from resected tumours, and inferior outcomes with anti-PD-1 antibodies (21). Mutations in several components of the Janus kinase (JAK1/JAK2) pathway have been implicated in both acquired (22) and primary (23) immune resistance in melanoma, by impairing interferon gamma (IFN-γ) signalling. Thus, screening for JAK1/2 mutations has been proposed as a mechanism to identify patients unlikely to respond to immune checkpoint inhibitors.
Recent studies have implicated loss of antigen presentation as a key mechanism of resistance to immune checkpoint inhibitors. β2microglobulin (β2M) is an essential component of MHC class I antigen presentation in which point mutations, deletions or loss of heterozygosity (LOH) have been identified in 30% of melanoma patients with progressing disease (24). In metastatic melanoma patients treated with anti-CTLA-4 and anti-PD-1 agents, β2M LOH was enriched threefold in non-responders compared to responders and was associated with poorer overall survival. Loss of both copies of β2M was found only in non-responders.
A further factor implicated in driving resistance to immune checkpoint inhibitors is transforming growth factor beta (TGF-β) (25). TGF-β is a multi-functional cytokine involved in the regulation of many cellular processes including cell proliferation, differentiation and survival. Melanoma produces increasing amounts of TGF-β with disease progression, inhibiting immune responses and providing an optimal microenvironment for undisturbed tumour growth. Its role as a response biomarker needs further investigation.
Many immune-based biomarker candidates have been identified to date in retrospective datasets, or preclinical models. The majority of these studies have focused on immune cells, either within the tumour, or circulating in blood.
Tumour-based immune-related biomarkers
The inflamed tumour microenvironment is characterized by the presence of T-cell markers and chemokines that mediate effector T-cell recruitment, with enhanced numbers of CD8+ T cells, macrophages, as well as some B cells and plasma cells. Therefore, it is perhaps not surprising that one of the most reproducible factors predicting response to immunotherapy in melanoma patients has been the presence of tumour-infiltrating lymphocytes (TILs) within tumours: increased numbers of TILs generally correlates with improved response and survival (26). Tumour infiltrating immune cells include T cells, macrophages and various types of immune suppressive cells, all of which contribute to the balance of a pro-immunogenic versus immunosuppressive microenvironment. Thus, low intratumoral CD8:CD4 ratios correlate with lack of response to treatment, while response rates as high as 80% have been reported to be associated with high intratumoral CD8:CD4 in metastatic melanoma patients treated with anti-PD-1 monotherapy (27). Because the nature of the immune microenvironment of a tumour at baseline is associated with efficacy of immune checkpoint inhibition, the assessment of an individual’s immune signature to predict treatment outcome is an area of active investigation. This emerging concept, known as immunoprofiling, relies on the ‘immunoscore’: an assessment of the type, density, and location of immune cells (28). Absolute numbers is a gross oversimplification of a highly complex microenvironment influencing T-cell function. It is likely that multiple markers may need to be combined to fully encompass the heterogeneity of immune cell responses in individual patients receiving specific therapies.
One way of combining multiple factors affecting response to immunotherapy is by gene expression profiling of tumour tissue. A T-cell inflamed tumour microenvironment rich in pro-inflammatory chemokines with an IFN-γ signature has been shown to correlate with the clinical efficacy of immune checkpoint inhibitors in melanoma patients (29–31). Several multi-gene expression profiles have been proposed as having predictive value, although results are not always consistent across studies. How-ever, evidence from a large cohort of > 300 tumours from multiple cancers including melanoma reported that integrated analysis of an immune gene signature combined with TMB enriches for anti-PD1 responders (32) (Fig. 4). This novel approach may provide a precision medicine framework for stratifying patient therapy in the future.
Fig. 4. Biomarker-defined responses to pembrolizumab monotherapy identify targetable resistance biology. (A) Tumours have low TMB and low neoantigenicity and lack a T cell-inflamed TME. (B) Tumours can evade the immune response despite high TMB and high neoantigenicity. (C) Although T cells are present, stromal and/or endothelial factors in the TME, low TMB and low neoantigenicity impede their activity. (D) Tumours have high TMB, high neoantigenicity and a T cell-inflamed TME, typified by activated T cells and other immune cells with cytolytic roles. (From Cristescu R et al., Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018 Oct 12;362(6411). pii: eaar3593. doi: 10.1126/science.aar3593. Reprinted with permission from The American Association for the Advancement of Science).
Blood-based biomarkers
Multiple blood-based biomarkers have been identified in retrospective studies and show promise to predict both response, and, potentially, toxicity, and have been extensively reviewed elsewhere (33–35). They include absolute neutrophil count, absolute lymphocyte count, neutrophil:lymphocyte ratio, absolute eosinophil count, relative lymphocyte count (RLC), absolute monocyte count, antibodies against NY-ESO1, T-regulatory cell count, and myeloid-derived suppressor cell (MDSC) count. Recent analysis of patients recruited to the CheckMate 064, 066 and 067 trials identified serum IL6 and CRP as predictors of improved response and survival after checkpoint blockade (36). Even so, most studies have been undertaken on small cohorts using a variety of different evaluation criteria (37) and all require validation in larger prospective trials.
The most extensive analysis of the effects of immune checkpoint inhibitors on peripheral blood was performed in metastatic melanoma patients treated with pembrolizumab (38). The study showed that 1) PD1 inhibition leads to an on-target immunological effect on CD8 T cells and this effect can be detected, longitudinally monitored and mechanistically interrogated in the peripheral blood with the major cell type affected being the Ki67+ CD8 T-cell population, characteristic of exhausted T cells (Tex). 2) Most patients had a single peak of anti-PD-1-induced immune reinvigoration, despite on-going treatment which occurred early during treatment (within 3–6 weeks). 3) Since the Tex cells were the major target of PD-1 blockade in most patients, the authors were able to develop a ‘reinvigoration score’ by relating changes in circulating Tex cells to tumour burden. 4) Responding Tex cells in the blood contained T-cell receptor clones shared with tumour-infiltrating T cells, and 5) The ratio of Tex-cell reinvigoration to tumour burden distinguished clinical outcomes and predicted for response. The relationship between Tex-cell reinvigoration and tumour burden suggests a ‘calibration’ of immune responses to antigen burden and raises the possibility that even robust reinvigoration by anti-PD-1 therapy may be clinically ineffective if the tumour burden is high. This study provides a clinically accessible potential on-treatment predictor of response to PD-1 blockade which now needs validating prospectively.
There are now several mature technologies available for plasma and serum protein identification and quantification, including mass spectrometry proteome profiling and affinity-based methods (37), which offer the opportunity for larger scale analyses and have identified several potential protein-based biomarkers. They include vascular endothelial growth factor (VEGF). Since an early observation that high serum VEGF were associated with decreased overall survival in metastatic melanoma patients treated with ipi (39), angiogenesis is increasingly appreciated as an immune modulator with therapeutic potential combined with checkpoint blockade. Markers of angiogenesis are now receiving increasing attention for their potential clinical application.
Changes in IL-17, CD8 T-cell clonal expansion, eosinophil counts, and markers of neutrophil activation have been associated with specific immune-related adverse events (irAEs) after treatment induction, but did not predict toxicity development when tested at baseline (40–42). Several other potential baseline risk factors for development of irAEs from ICPIs have been suggested, including a family history of autoimmune diseases (43, 44), but these require further validation. It is intriguing to suggest that similar genetic loci that predispose to autoimmune conditions also contribute towards development of irAES but, to date, no germline factors have been associated with development of irAEs (45). Similarly, preliminary studies suggest the microbiome (discussed in more detail below) may influence risk of irAEs, particularly colitis (46).
A recent study implicated a group of cytokines in predicting immune checkpoint mediated toxicity (47). Eleven cytokines (including pro-inflammatory cytokines such as IL-1a, IL-2 and IFNa2; developed into a score called the ‘CYTOX score’) measured both pre- and early during treatment were found to be significantly up-regulated in patients with severe immune-related toxicities in 98 melanoma patients treated with PD-1 inhibitors, alone or in combination with anti-CTLA-4. The findings were then validated in an independent validation cohort of 49 patients treated with combination anti-PD-1 and anti-CTLA-4. If validated in larger prospective studies, the CYTOX score could identify toxicity-prone patients to either avoid harmful treatment or consider prophylactic interventions to mitigate side effects.
The microbiome
The gut microbiome influences host immunity and has been implicated in multiple diseases including cancer. The presence of certain gut bacteria, including Akkermansia muciniphila and Bifidobacterium, was reported to improve efficacy of PD-1 blockade in animal models. In melanoma patients, significant differences have been reported in the composition and diversity of the gut microbiome between responders and non-responders to anti–PD-1 immunotherapy. However, the reported findings have so far been inconsistent (48–52), which may say more about the limitations of the sequencing technology being used. Even so, the significance of the microbiome is further implicated by preliminary studies suggesting that antibiotic (53, 54), probiotic and prebiotic (ie. dietary fibre) intake all can all influence response to checkpoint inhibition.
A key element of drug development is understanding drug-induced toxicity, whether on-target or off-target effects, and whether toxicity has any correlation with predicting efficacy. In the context of BRAF-targeted agents, there is no evidence that the two are connected. With checkpoint inhibitors, the data is far more intriguing, although not at all clear cut. For ipilimumab, immune-related adverse events do not correlate with response, or survival (55, 56). For anti-PD-1 monotherapy, results are conflicting, both in the advanced (57, 58), and most recently in the adjuvant setting (59, 60). The most compelling data comes from the CheckMate 067 trial, when it was observed that 68% of patients receiving combination ipi+nivo who stopped treatment early due to unacceptable toxicity continued to maintain a response over time (15). Thus, at least for metastatic melanoma patients receiving ipi+nivo, it is reasonable to reassure patients experiencing severe, sometimes life-threatening toxicity, that this may predict for good outcome, although the converse is not necessarily true. Understanding the mechanisms that underlie irAEs and their optimal management are key areas requiring active research.
Anti-PD-1 antibodies are licensed to be administered to metastatic melanoma patients for as long as there is evidence of clinical benefit. For those patients who respond, they may be consigned to treatment for many years, risking toxicity, impacting quality of life, and requiring significant healthcare resources. Adjuvant therapy has been approved for a duration of one year. The biological necessity for long term therapy in either setting is not determined and in fact, there is accumulating evidence arguing against the need. Evidence from following-up advanced melanoma patients stopping treatment due to toxicity suggest that response can be maintained in the absence of drug being administered. Long term follow-up of melanoma patients recruited to the KEYNOTE 006 trial who stopped treatment after 2 years reported durable complete remissions after discontinuation and low incidence of relapse (61). The mechanisms underlying this observation clearly need to be studied, but functional imaging may be a useful adjuvant to clinical decision-making.
Retrospective data from 104 metastatic melanoma patients treated with anti-PD1 antibodies suggests that performing 18F-2-fluoro-2-deoxy-D-glucose positron emission tomography (FDG-PET) at one year accurately predicts long-term outcome: PFS of complete metabolic response (CMR) was 96%, compared with 49% without CMR (HR 0.06, p<0.06) (62). The UK DANTE study is randomizing melanoma patients who are progression-free after one year of anti-PD1 antibody therapy to either stop or continue treatment. A sub-study has been proposed to evaluate prospectively the value of performing PET at one year and will also determine the value of earlier PET scanning performed at or before the first routine 12 week CT response assessment. The rationale for shorter duration of adjuvant therapy also warrants evaluation in randomised trials.
Now that systemic therapy is established for treatment of both metastatic and high-risk resected melanoma, a key next phase of research is to optimize selection of treatment by identifying biomarkers which can reliably predict both response to and relapse on therapy. This rapidly evolving and expanding personalized approach, offers the opportunity safer, more cost-effective healthcare in years to come.


 Click to show fullsize
Click to show fullsize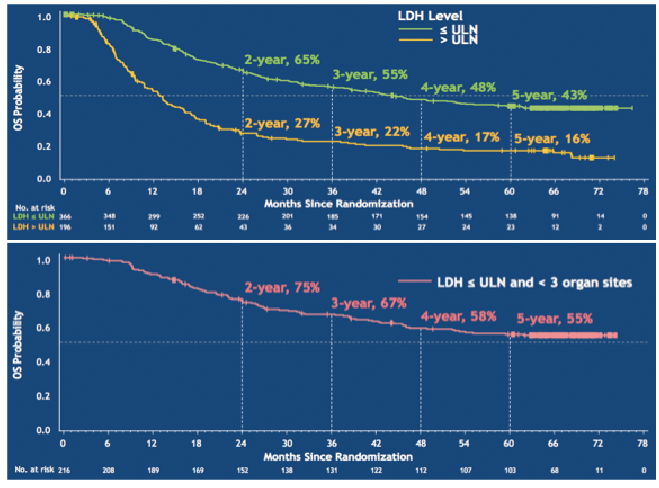 Click to show fullsize
Click to show fullsize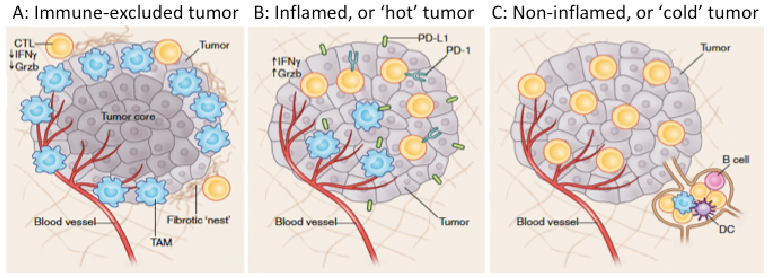 Click to show fullsize
Click to show fullsize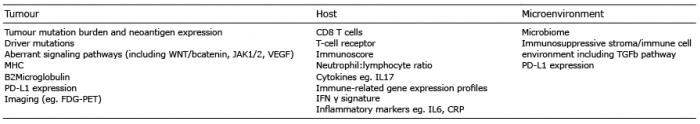 Click to show fullsize
Click to show fullsize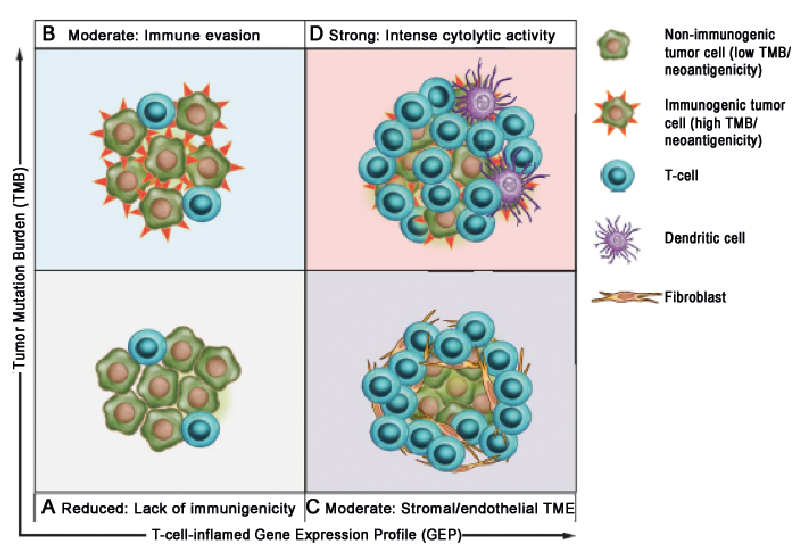 Click to show fullsize
Click to show fullsize