


Departments of 1Dermatology, Venereology and Allergology, 2Biology and Medical Genetics, 3Pathology, 4Paediatrics, Paediatric Gastroenterology, Allergology and Nutrition, and 5Allergology, Medical University of Gda?sk, 17 Smoluchowskiego St, PL-80-214 Gda?sk, Poland. *E-mail: m.lange@gumed.edu.pl
Accepted Apr 28, 2020; Epub ahead of print May 4, 2020
Acta Derm Venereol 2020; 100: adv00149
Mastocytosis is a heterogeneous disease that results from a clonal, neoplastic proliferation of morphologically and immunophenotypically abnormal mast cells (MCs), which accumulate in one or more organ systems (1, 2). Paediatric mastocytosis is considered a benign, transient skin-limited disease, defined as cutaneous mastocytosis (CM) (3, 4). However, analysis of the literature data on childhood-onset mastocytosis shows that systemic mastocytosis (SM) is a rare finding in children (5–9). At present, the true incidence of SM in children remains unknown. According to the European Competence Network on Mastocytosis (ECNM) diagnostic recommendations, in children with cutaneous manifestation of mastocytosis, a bone marrow (BM) investigation is not necessary unless serum tryptase levels are very high (> 100 ng/ml) or the blood count and/or other clinical findings and symptoms suggest the presence of a haematological neoplasm (10). Currently, the use of highly sensitive PCR techniques to identify the KIT D816V mutation in peripheral blood (PB) has been incorporated into the algorithm for diagnosis of SM, both in adults and children (6, 11–13). The current study aimed to assess whether detection of the KIT D816V mutation in PB is a valuable tool for diagnosis of SM in children with extensive skin lesions in daily clinical practice.
A total of 32 children, age range 1–14 years, presented with diffuse CM (DCM) (n = 16) or maculopapular CM (MPCM) involving over 50% of BSA (n = 16), were enrolled in the study, following informed consent. The study was approved by the local medical research ethics committee. The population consisted of 18 (56%) males and 14 (44%) females, who regularly attended our mastocytosis centre. The median age at the initial evaluation at our centre was 2 years (range 1–14 years). The median follow-up was 6 years (range 2–11 years). Patients were given diagnoses and classified into different disease categories according to established criteria (1–3). Genomic DNA isolated from PB of all patients was used for molecular studies. Mutational analysis of KIT D816V was performed using a TaqMan Universal PCR Master Mix with an AmpErase UNG (Thermo Fisher Scientific and Light Cycler 480 II (Roche) assay according to the recommendations described in detail by Kristensen et al. (11, 12). The assay is considered positive, with values of > 0.01% (6). For statistical analysis, the STATISTICA v 12.5 (Statsoft) program was used. All p-values were considered to be statistically significant if less than 0.05.
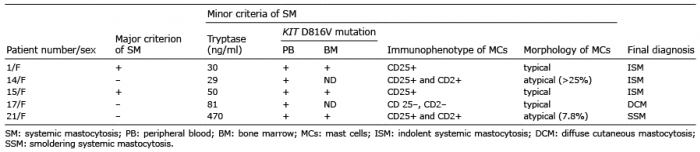
Thirty-two paediatric patients presenting with DCM (6 females and 10 males) and extensive MPCM (8 females and 8 males) were diagnosed to check whether they are at risk of SM. The results of routine laboratory studies (including full blood count and a differential, and biochemistry) were within the normal ranges in the majority of children, and those with abnormal indices were transient and had no clinical significance. Organomegaly was found in abdominal ultrasounds in 6 (19%) of 32 children (hepatomegaly n = 4, hepatosplenomegaly n = 1, splenomegaly n = 1). All patients with organomegaly had no palpable liver and/or spleen below the costal margin, and no clinical and laboratory features of impairment of liver and/or spleen function in the monitoring period. The determination of serum tryptase levels at the first evaluation revealed that in 28 (87.5%) of 32 children, the levels were over the normal ranges (> 11.4 ng/ml). The PB KIT D816V assay was positive in 11 (34%) of overall 32 children, including DCM (n = 4) and MPCM (n = 7) subjects. Five KIT D816V-positive children with high suspicion of SM underwent further BM investigation (Table I). All of them had persistently elevated or rising serum tryptase levels, ranging from 29 to 470 ng/ml. Hepatomegaly was found in one patient (patient number 21). A lack of regression of skin lesions in patients who were over 18 years of age during follow-up was observed in 2 patients (numbers 1 and 15). Finally, SM was diagnosed in 4 patients, representing 12.5% of the overall 32 children enrolled in the study and 36.4% of KIT D816V-positive subjects. The diagnosis of SM was established based on the fulfilment of one major criterion and 3 minor criteria (n = 2) or at least 3 minor criteria (n = 2). Three children were categorized as indolent SM (ISM). One SM child (patient number 21) displayed two B findings, such as serum tryptase levels > 200 ng/ml and hepatomegaly without impairment of liver function, and therefore smoldering form (SSM) of SM was diagnosed (2). The remaining KIT D816V-positive children (n = 7) and all KIT D816V-negative children (n = 21) did not require further diagnostic procedures at the last evaluation because of the decrease in serum tryptase levels in the follow-up, the lack of organomegaly, and the lack of clinically significant abnormalities in the complete blood count. Therefore, 28 of 32 (87.5%) children were categorized as CM. The KIT D816V mutation in PB was found in all patients with SM (n = 4) and in 25% (n = 7) of those with CM, which indicates that the mutation was detected more commonly in children with SM than in those with CM (p = 0.009). The sensitivity of the PB KIT D816V assay was 100% (95% confidence interval (95% CI) 39.58–100), and the specificity was 75% (95% CI 54.78–88.57) when calculated for the entire group of 32 patients.
Table I. Diagnostic criteria in children with highly suspected systemic mastocytosis
This paper presents the results of a study on the diagnostic value of the KIT D816V mutation in PB in a selected group of children with mastocytosis presenting with extensive skin involvement. So far, there is only one published study in which the KIT D816V mutation was determined in the PB of children (6). In that study, Carter et al. proved that the detection of this mutation in PB strongly suggests systemic disease (6). At this point, our observations are in line with their findings. In contrast to our selected study group, those authors enrolled children with all forms of CM, and ISM, and a probable diagnosis of SM (6). The limitations of the current study are that it was a single-centre study and the relatively small number of patients enrolled due to the very rare occurrence of DCM and severe CM involving over 50% of BSA. Our results suggest that determination of the KIT D816V mutation in PB is of great importance to identify the subgroup of children with mastocytosis who are at risk of SM. Therefore, CM children with positive KIT D816V mutation in PB and moderately elevated serum tryptase level require more careful follow-up and further BM investigation in selected cases. In our opinion, BM biopsy is indicated in CM children with KIT D816V mutation in PB when they have increasing serum tryptase over time, clinically significant abnormalities in blood count, or organomegaly in abdominal ultrasound. So far, measurement of serum tryptase levels was the only non-invasive procedure involved in the diagnostic work-up and monitoring of the course of paediatric mastocytosis (7–10). Nowadays, the determination of the KIT D816V mutation in PB is another non-invasive, procedure that has been proposed for the same purposes in children (6).
In conclusion, the results of the current study prove that determination of the KIT D816V mutation in PB is a useful diagnostic tool in daily clinical practice for children with extensive skin involvement.
Funding. Polish Ministry of Science and Higher Education grant 02-0066/07/253 and grant 02-0060/07/153
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize