


1Department of Dermatology and 3Department of Rheumatology Hospital, Universitario Virgen de las Nieves, and 2Instituto de Investigacion Biosanitaria ibs.Granada, Granada, Spain
The aim of this study was to determine the prevalence and type of cutaneous manifestations which occur in patients with granulomatosis with polyangiitis and to explore the potential association between cutaneous and systemic involvement in these patients. A retrospective case series study was designed, including all granulomatosis with polyangiitis cases diagnosed between 2010 and 2018 at the Hospital Universitario Virgen de las Nieves. Thirty-nine patients with granulomatosis with polyangiitis were identified, of which 53.85% presented cutaneous manifestations. In decreasing order of frequency, the types of cutaneous problems observed included: palpable purpura, muco-cutaneous ulcers, subcutaneous nodules, pyoderma gangrenosum-like ulcers, digital necrosis, papulo-necrotic lesions and livedo reticularis. Patients with palp-able purpura presented a higher frequency of renal involvement (p = 0.008). Cutaneous manifestations of granulomatosis with polyangiitis may facilitate early disease diagnosis. Likewise, a manifestation such as palpable purpura may be a predictor of kidney damage.
Key words: granulomatosis with polyangiitis; skin; vascular purpura.
Accepted May 4, 2020; Epub ahead of print May 6, 2020
Acta Derm Venereol 2020; 100: adv00150.
Corr: Alejandro Molina-Leyva, Department of Dermatology, Hospital Universitario Virgen de las Nieves. Avenida de Madrid, 15, CP 18012, Granada, Spain. E-mail: alejandromolinaleyva@gmail.com
A specific skin finding has not previously been linked to the presence of systemic involvement in granulomatosis with polyangiitis. We studied 39 patients diagnosed with granulomatosis with polyangiitis, 53.85% cases presented cutaneous manifestations. We observed several types of cutaneous problems: palpable purpura, mucocutaneous ulcers, subcutaneous nodules, pyoderma gangrenosum-like ulcers, digital necrosis, papulonecrotic lesions and livedo reticularis. Moreover, we found that patients with palpable purpura presented a higher frequency of renal involvement. In that way, cutaneous manifestations of granulomatosis with polyangiitis may facilitate an early diagnosis and a specific skin finding, the palpable purpura, could be a predictor of kidney damage.
Granulomatosis with polyangiitis (GPA), previously known as Wegener granulomatosis (1), is a systemic vasculitis included in the ANCA vasculitis group which usually affects small to medium vessels (2, 3). GPA is a relatively rare condition. Its prevalence is estimated at between 5 and 12 cases per 100,000 population (4). The estimated age of onset ranges between 45 and 65 years old. No difference between sexes has been reported (5).
Its pathogenesis remains unclear but current evidence suggests that GPA may develop as a result of complex gene-environment interactions (6). It has been related to dust (specifically silica and grain dust) and infections associated with farming (7). Prognosis without treatment is poor, with a patient mortality rate of 82% within the first year (8).
GPA is associated with the presence of ANCA, which usually presents a cytoplasmic pattern in immunofluorescence and antibodies directed against PR3 in the ELISA test. This finding is the most specific and sensitive criteria for diagnosis (9, 10). However, a biopsy should be performed to confirm the diagnosis (11), and any affected organ can be biopsied. Several histopathological entities are characteristic of cutaneous GPA, such as leukocytoclastic vasculitis and necrotizing granulomatous inflammation (12, 13).
GPA can involve any organ, but it predominantly affects the upper and lower respiratory tract (90%), kidneys (60%) and eyes (40%) (14). Less commonly, it can also produce neurological, gastrointestinal or cardiological damage. Kidney damage worsens GPA prognosis (16): the 10-year survival rate is estimated at 40% when the kidneys are involved and 60–70% when there is no renal involvement (10). The prevalence of cutaneous manifestation ranges between 35 and 50%. Cutaneous lesions may be present at disease onset (12, 13). Palpable purpura has been described as the most frequent skin lesion, although a wide array of clinical features may be observed (14–28).
First-line treatment recommendations for GPA include the combination of glucocorticoids and cyclophosphamide, which achieves disease control in up to 75% of patients and a 90% survival rate (29). Recent data suggests that corticosteroids combined with rituximab could be equally effective (30). Second- and third-line drugs are trimethoprim–sulfamethoxazole (for the limited form of GPA), azathioprine and methotrexate (18).
The objectives of this study are: (i) to determine the prevalence and type of cutaneous manifestations which occur in patients with GPA, and (ii) to explore the potential association between cutaneous and systemic involvement in patients with GPA.
Design: Retrospective case series study including all GPA patients diagnosed between 2010 and 2018 at the Hospital Universitario Virgen de las Nieves, Granada (Spain). The cases were identified through digital medical records. This study was approved by the ethics committee of the Hospital Universitario Virgen de las Nieves.
Inclusion criteria: patients diagnosed with GPA according to the European League Against Rheumatism (EULAR) guidelines (31, 32). At least 3 of these 6 criteria are required for GPA diagnosis: 1) upper tract respiratory involvement, 2) typical lesions on chest x-ray or CT, 3) alterations in urinalysis, 4) biopsy with granulomatous inflammation, 5) airway stenosis, 6) serological alterations (anti-PR3, ANCAs).
Exclusion criteria: patients with incomplete data for any of the variables of interest and those who did not meet EULAR GPA criteria.
The main variables of interest were sex, current age, age at diagnosis, location and type of cutaneous lesions, systemic organ involvement, ANCA status at disease onset, treatment used, outcome and histopathologic characteristics. Renal involvement was defined by a proteinuria of more than 0.5g/d and/or haematuria.
Statistical analysis: Descriptive statistics were used to explore the sample characteristics. Continuous data is expressed as the median (interquartile range). The absolute and relative frequency distributions were estimated for qualitative variables. The Shapiro-Wilk test was used to check the normality of the variables. The Mann-Whitney U test was used to compare continuous nonparametric variables. The χ2 test or Fisher’s exact test were applied to nominal data, where necessary. Significance was set at two tails (p < 0.05) for all tests. Statistical Analyses were performed using SPSS version 22.0 (IBM Corp, Armonk, NY, USA).
Clinical and demographic characteristics
Thirty-nine patients (19 men and 20 women) diagnosed with GPA were identified, aged between 18 and 77 years old. The median duration of the disease was 6 years (range 3–12) . Twenty-one cases (10 men and 11 women) out of 39 (53.85%) showed cutaneous manifestations. Only 5 patients (5/29, 12.82%) presented cutaneous involvement at disease onset.
Table SI shows the characteristics of patients with cutaneous manifestations. Regarding patients with cutaneous involvement, 71.43% (15/21 patients) also had upper respiratory tract involvement, 52.38% (11/21) had pulmonary damage and 47.62% (10/21) had renal involvement. Systemic involvement also included ocular (4/21, 19.05%), peripheral nervous system (4/21, 19.05%) and cardiovascular system (3/21, 14.29%) manifestations.
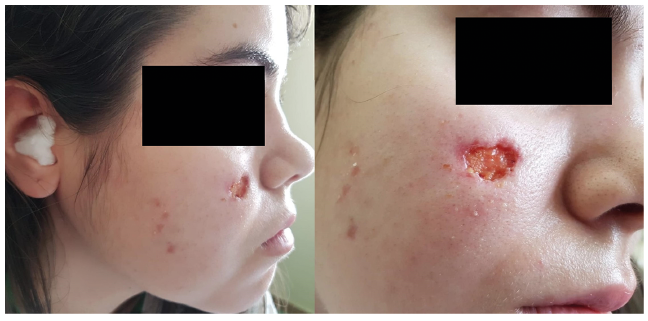
Cutaneous lesions in our case series included palpable purpura (8/21, 38.10%), mucocutaneous ulcers (6/21, 28.58%) (Fig. 1), subcutaneous nodules (3/21, 14.21%), pyoderma gangrenosum-like ulcers (2/21, 9.52%) (Fig. 2), digital necrosis (2/21, 9.52%) (Fig. 2), papulonecrotic lesions (1/21, 4.76%) and livedo reticularis (1/21, 4.76%). Two patients presented several types of cutaneous manifestations. The most frequently involved sites were the lower limbs (12/21, 57.14%), followed by the face (5/21, 23.81%).
Fig. 1. Saddle nose deformity and a facial ulcer as the initial presentation of granulomatosis with polyangiitis in a woman in her 20s.
Fig. 2. A pyoderma gangrenosum-like ulcers and digital necrosis in a woman in her 50s with granulomatosis with polyangiitis.
Histopathologic characteristics
A skin biopsy specimen was obtained from 9 patients. Some of the patients had been diagnosed with GPA before cutaneous manifestation appeared and other patients were diagnosed by taking a biopsy from a different affected organ. In cases 3, 4 and 6, the histologic findings showed features of leukocytoclastic vasculitis. In case 13, the biopsy specimen results revealed ulceration with metaplasia and granulation tissue. In case 15, the histologic findings showed fragments of nasal mucosa without relevant alterations. In case 16, a nasal biopsy showed granulomatous rinosinusitis with thrombotic vasculitis. In case 18, the histologic findings were intense inflammatory histiocytary reaction with involvement in small and medium-size vascular walls (angiitis with partial necrosis) and formation of intraluminal fibrin. In case 19, the nasal mucosa biopsy results showed a chronic ulcer with granulation tissue and nonspecific inflammation. In case 21, the histologic features of the lesions included granulomatous and inflammation with follicular destruction.
Treatment and follow-up
All patients were treated with systemic glucocorticoids, 28.2% (11/39) with cyclophosphamide, 25.6% (10/39) with methotrexate, 20.5% (8/39) with azathioprine, 17.9% (7/39) with trimethoprim–sulfamethoxazole, 15.4% (6/39) with mycophenolate and 12.8% (5/39) with rituximab. One patient was also treated with tocilizumab because he also suffered from rheumatoid arthritis. Four patients needed more than 2 immunosuppressive agents to control the disease.
The follow-up periods ranged from 1 to 15 years (median 6, interquartile range 3–12 years). Twenty out of 39 patients (51.3%) remained in remission at the end of follow-up, 4 patients (10.3%) had uncontrolled disease despite treatment and 3 patients (7.7%) died during the follow-up. Follow-up was limited in 12 patients (30.8%).
Implications of cutaneous involvement
Table SII shows the characteristics of patients with GPA, according to the presence of cutaneous manifestations. Patients with cutaneous manifestations showed a trend towards lower frequency of lung damage (p = 0.09).
Afterwards, analyses of the specific types of lesions and systemic manifestations were carried out. No association was found regarding mucocutaneous ulcers, subcutaneous nodules, pyoderma gangrenosum-like ulcers, digital necrosis, papulonecrotic lesions or livedo reticularis. Differences were found between patients with and without palpable purpura (Table SIII). Palpable purpura was more frequent in men (p = 0.024). An association between palpable purpura and renal involvement was noted. Patients with palpable purpura presented a higher frequency of renal involvement (p = 0.008). Six out of 7 patients (85.71%) presented palpable purpura preceding the onset of kidney damage. Renal involvement developed 6 to 34 days after palpable purpura. Three patients with palpable purpura needed more than two immunosuppressive agents to control the disease.
The results of this study show that patients with GPA frequently present cutaneous involvement during disease evolution, but skin involvement is not usually present at disease onset. Palpable purpura was the most frequent cutaneous manifestation and the lower limbs were the most frequent location. The results of our study are consistent with other case series published, regarding the prevalence of cutaneous manifestation in GPA (19–23). Only one study identifies a lower frequency of cutaneous manifestations, 14% (34/244), and a higher frequency of cutaneous involvement at disease onset, 62% (21/34) (20).
In agreement with previous reports, we observed that palpable purpura was more frequent in men and its presence was associated with a higher frequency of renal involvement. Other cutaneous manifestations present in our case series were mucocutaneous ulcers, subcutaneous nodules, pyoderma gangrenosum-like ulcers, digital necrosis, papulonecrotic lesions and livedo reticularis. All of them had previously been reported as possible cutaneous manifestations in GPA (19–23). In our study, lower limbs were the most frequently involved site. One study reported that upper extremities were the most frequently involved site in paediatric cases of GPA (22). The location of skin lesions may vary depending on age.
Francés et al. (23). also explored the potential association of cutaneous manifestations with systemic involvement and concluded that cutaneous manifestations were potentially associated with a higher frequency of articular and renal involvement. In our study, we found greater frequency of renal involvement specifically in patients with palpable purpura. We found no association between other types of cutaneous manifestations and systemic symptoms although our sample size was limited. We did not find any previous studies analysing the relationship between the different types of cutaneous manifestations of GPA with another systemic involvement.
Palpable purpura could be a predictor of kidney damage in other forms of vasculitis. In Henoch–Schönlein purpura (HSP), skin involvement is caused by direct IgA1-immune deposits, and nephropathy is produced by immune complexes containing galactose-deficient (Gd-)IgA1 created after IgA1-immune deposits (33). A similar mechanism of pathogenesis mediated by ANCA could explain why palpable purpura may precede renal involvement in patients with GPA. In fact, in HSP, renal manifestations developed over a period of several days to one month after palpable purpura (34), a similar range as that described in our case series, although the median time between the appearance of cutaneous manifestation and the development of systemic involvement has not been described. There has only been one reported case of a 14-year-old girl with GPA who presented with palpable purpura two weeks before renal involvement (35). On the other hand, lung involvement appears to depend on T lymphocytes and granuloma formation (36). These different pathogenic mechanisms could explain why patients with cutaneous symptoms may have less frequent pulmonary involvement.
GPA can be a fatal condition. Kidney involvement is one of the main causes of mortality and kidney damage worsens the prognosis (37). The results of our study suggest that patients with palpable purpura could be at higher risk of renal involvement and should be carefully followed up. The appearance of palpable purpura could mark renal involvement and the need for treatment intensification in these patients. Prospective and larger multicentric studies are needed to confirm our findings.
The results of our study should be considered in light of certain methodological limitations: (i) Limited sample size. (ii) Retrospective study. (iii) Different follow-up times in some patients.
This study outlines the importance of the cutaneous manifestations of GPA that may facilitate early diagnosis of the disease. Likewise, a manifestation such as palpable purpura may be a predictor of kidney damage. These results might help physicians to monitor these patients more closely and initiate early enhanced treatment.


 Click to show fullsize
Click to show fullsize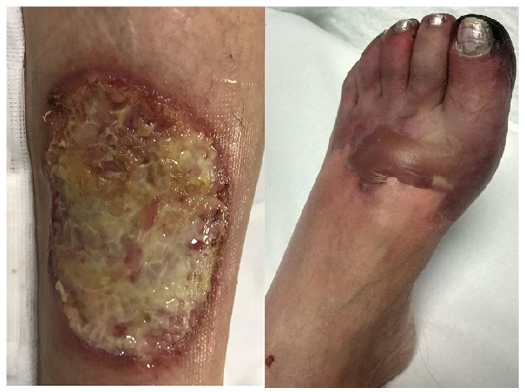 Click to show fullsize
Click to show fullsize