


Departments of 1Oncodermatology, 2Haematology and 3Biopathology, Reims University Hospital, Reims, Departments of Dermatology, 4Bordeaux University Hospital, Bordeaux, 5Department of Dermatology, Rennes University Hospital, Rennes, France, 6Inselspital Bern University Hospital, University of Bern, Switzerland, 7Avicenne Hospital, Bobigny, 9Lille University Hospital, Lille, 12Hospital of Cochin, Paris, 13Angers University Hospital, Angers, 14Tours University Hospital, Tours and 16Saint Louis Hospital, APHP, Paris, Departments of Biopathology, 8Hospital of Cochin, Paris, 11Lille University Hospital, Lille, 15Henri Mondor Hospital, Créteil and 17Bordeaux University Hospital, Bordeaux, and 10Department of Biology, Reims University Hospital, Reims, France
Cutaneous involvement in Waldenström’s macroglobulinaemia (WM) has been poorly characterized. To describe this involvement, a retrospective study of 19 patients with WM and cutaneous involvement of tumour B cells was performed. Twelve patients (group 1) had lymphoplasmacytic, non-transformed cutaneous proliferation, while in 7 cases (group 2) cutaneous involvement corresponded to histological transformation. In group 1, skin involvement was inaugural in 6 cases. The lesions were infiltrated plaques (83%), papules (25%) and tumours (42%). Four patients had a similar clinical picture (purplish, bilateral and symmetrical infiltration on the face). MYD88 L265P mutation was detected in the skin biopsy in all 6 cases tested. The 3-year specific survival rate was 88%. In group 2, cutaneous transformation occurred during the follow-up of the WM (71%). Lesions presented as ulcerated tumours (86%) of the trunk (57%) and lower limbs (57%). The 3-year specific survival rate was 22%. Skin involvement in WM has distinctive characteristics (e.g. clinical, histological, immunohistochemical, MYD88 L265P mutation).
Key words: Waldenström’s macroglobulinaemia; cutaneous lymphoma; diffuse large B-cell lymphoma; histological transformation; extranodal involvement; MYD88 L265P mutation.
Accepted May 19, 2020; Epub ahead of print May 25, 2020
Acta Derm Venereol 2020; 100: adv00225.
Corr: Sarah Stien, Department of Oncodermatology, Robert Debré Hospital, avenue du Général Koenig, FR-51092 Reims Cedex, France. E-mail: sarah.stien@gmail.com
Skin involvement in Waldenström’s macroglobulinaemia is poorly described, and diagnosis of this disorder may be difficult. Many differential diagnoses are possible (other lymphomas, primary cutaneous lymphoma, etc.). Describing 2 aspects of Waldenström’s macroglobulinaemia (infiltration by lymphoplasmacytic tumour cells or histological transformation into diffuse large B-cell lymphoma) allows the identification of clinical, histological and immunohistochemical diagnostic aids. Detection of the MYD88 L265P mutation is of interest. This study identified a characteristic clinical picture in non-transformed cases, and was able to distinguish between transformed and non-transformed cases by their severity and poor prognosis.
Waldenström’s macroglobulinaemia (WM) is a rare B-cell lymphoproliferative disorder characterized by lymphoplasmacytic bone marrow infiltration and the presence of a serum monoclonal immunoglobulin (Ig)M paraprotein. WM accounts for 1–2% of haematological malignancies (1, 2). Cutaneous lesions specific to WM include cutaneous macroglobulinosis and cutaneous involvement of neoplastic cells. Cutaneous macroglobulinosis results from cutaneous IgM deposits, leading to IgM storage papules (3). Cutaneous involvement of neo-plastic cells includes dermal infiltration by lymphoplasma-cytic tumour cells and/or histological transformation to diffuse large B-cell lymphoma (DLBCL) involving the skin (4). Cutaneous macroglobulinosis has been widely described (3). In contrast, only isolated case reports and small series of lymphomatous skin involvement associated with WM have been reported to date (5–7). The aim of this multicentre retrospective study was to describe clinical, biological and histological features, therapeutic approaches, clinical outcome and prognosis of cutaneous involvement in WM.
Identification of cases and inclusion criteria
The database of the French Cutaneous Lymphoma Study Group (GFELC) and the participating referring clinicians were queried to identify cases (recruitment method 1). Furthermore, cases with skin involvement previously included in a series of patients with a WM histologically transformed to DLBCL were also selected (recruitment method 2) (4). All cases were analysed for the following inclusion criteria: (i) a diagnosis of WM confirmed by a haematologist, based on criteria established in the Second International Workshop on Waldenström’s macroglobulinaemia (8); (ii) presence at any time of skin lesions diagnosed as cutaneous location of B-cell lymphoma, whatever the subtype. Exclusion criteria were: (i) age under 18 years; (ii) non-lymphomatous cutaneous lesions including cutaneous macroglobulinosis, and variable inflammatory, non-lymphomatous disorders after examination by GFELC pathologists; (iii) absence of informative clinical and pathological data available in medical records.
Informed consent was obtained for each patient. Patient records were anonymized prior to analysis. The database was built in accordance with protocol MR004 from the Commission Nationale de l’Informatique et des Libertés (number 2206749, 13/09/2018), and followed the requirements of the French authorities.
Collected data
The following data were collected at the time of WM and cutaneous involvement: age; sex; Eastern Cooperative Oncology Group (ECOG) performance status (PS); type (papules, plaques, tumours, ulcerated or not), colour and anatomical location of skin lesions, based on medical reports and available pictures; clinical symptoms (pain and pruritus); presence of lymphadenopathy; presence of extranodal non-cutaneous involvement; complete blood counts; serum monoclonal IgM; serum lactate dehydrogenase (LDH) and B2-microglobulin levels; the presence of MYD88 L265P mutation, detected by skin and/or blood analysis; histological and immunohistochemical characteristics of cutaneous lesions, as described on pathology reports; prognostic score, according to the International Prognostic Scoring System (IPSS) for WM, and according to the revised International Prognostic Index (R-IPI) for transformed WM; and follow-up data, including treatments, cutaneous and haematological response, date of last follow-up and status at last follow-up.
Statistical analysis
Quantitative variables were expressed as median and extreme values and qualitative variables as numbers and percentages. Specific survival was defined as the time from diagnosis of cutaneous involvement to death related to WM, or last follow-up. Death from other causes was considered censoring. Survival curves were plotted using the Kaplan–Meier method. Two groups were analysed separately: patients without histological transformation (group 1); and patients with histological transformation into DLBCL (group 2).
Using the first recruitment method, 12 patients were included in group 1, and 2 patients in group 2. The second recruitment method identified 5 cases of histological transformation WM that could be included in group 2. A total of 12 patients were included in group 1 and 7 in group 2.
Among patients in group 1, the skin lesions preceded the diagnosis of WM in 2 cases, by 3 and 7 years, respectively. In this last patient, an initial diagnosis of marginal zone lymphoma (MZL) had been made, and secondarily corrected to WM after the haematological diagnosis. In 6 cases, the WM preceded the occurrence of cutaneous lesions with a median delay of 3.5 years (1–16). In these cases, cutaneous progression was isolated at occurrence of skin lesions in 3 patients, while in 3 other cases there was an associated haematological progression. In 4 cases cutaneous, lesions were concomitant to the diagnosis of WM and therefore revealed the disease.
In group 2, skin involvement preceded the diagnosis of WM in one case, thus revealing the disease. In the other 6 cases, cutaneous involvement followed the diagnosis of WM with a median delay of 4 years (0.5–10).
Patients’ characteristics at cutaneous involvement
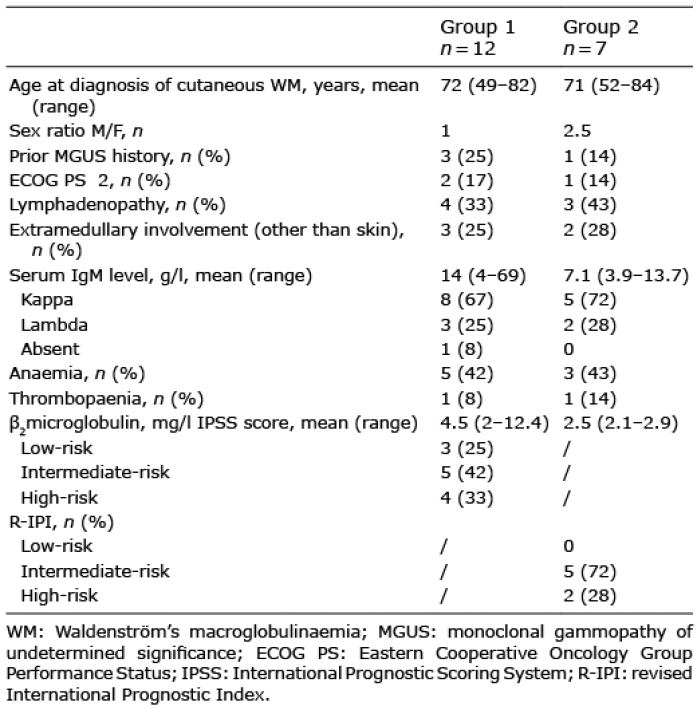
There were 12 men and 7 women, median age 70.5 years (range 48–84 years). Three patients (16%) had an ECOG PS ≥2 and 7 patients (37%) had lymphadenopathy. Extranodal, non-cutaneous involvement was present in 5 cases (26%): 3 cases in group 1 (with rectal, ocular, and adrenal and bone involvement, respectively), and 2 cases in group 2 (with adrenal and bone involvement). The median level of serum monoclonal IgM was 10 g/l (3.9–69 g/l) with kappa light chain in 13 cases and lambda light chain in 5 cases. One patient in group 1 was in haematological remission, without detectable serum monoclonal IgM at the time of skin involvement. Eight patients (42%) had anaemia and 2 (10.5%) had thrombopaenia. Serum LDH and ß2-microglobulin levels were elevated in 6 (31.5%) and 7 (37%) cases, respectively. The median ß2-microglobulin level was 4.3 mg/l (2–12.4). In group 1, according to the IPSS, 3 patients (25%) were classified as low-risk, 5 (42%) as intermediate-risk and 4 (33%) as high-risk. In group 2, according to the R-IPI, 5 patients (72%) were classified as intermediate-risk and 2 (28%) as high-risk. The characteristics of patients in each group are detailed separately in Table I, showing no significant difference in demographic and haematological features at the time of cutaneous involvement.
Table I. Patients’ characteristics at cutaneous involvement
Clinical characteristics of cutaneous lesions
In group 1, the clinical aspect could include several types of skin lesions in the same patient. The most common aspect was purplish, pink to copper-red infiltrated plaques (83%), papules (25%) and tumours (42%). Fifty percent of patients had lesions on the face, 42% had lesions on the lower limbs, 25% on the upper limbs and 25% on the trunk (Table II). Lesions were symmetrical and bilateral in 42% of cases. Four patients had a very similar clinical appearance, with purplish, bilateral and symmetrical infiltration of the cheekbones and ears (Fig. 1). Two of these patients also had lesions on the lower limbs. One patient reported a pruritus and 2 patients of painful skin lesions, while the other 9 patients had no symptoms related to the skin involvement. Lesions were ulcerated in only one case.
In group 2, the most common aspect was tumours (86%), often ulcerated (43%), involving the trunk (57%), the upper limbs (43%) and/or the lower limbs (57%).
Table II. Clinical characteristics of cutaneous involvement
Fig. 1. Four patients with similar clinical picture, with purplish, bilateral and symmetrical infiltration of the face (cheeks, nose, forehead and ears), corresponding to cutaneous involvement in non-transformed Waldenström’s macroglobulinaemia. Permission received from the patients to publish the photos.
Histological and immunohistochemical characteristics of cutaneous lesions
Skin biopsies of 12 patients in group 1 showed, under a normal epidermis, a dermal infiltrate made of small lymphocytes and plasma cells (10/12), or lymphoplasmocytoid cells (4/12), with a nodular (2/12), diffuse (5/12) or interstitial (2/12) pattern, sometimes perivascular (4/12) or periadnexal (2/12). The cutaneous lymphoplasmacytic infiltrate was positive for CD20 (12/12) and BCL2 (5/7), and negative for CD10 (5/5), BCL6 (5/5) and CD23 (3/3). Reactive CD-3 and CD5-positive T cells were often present. The Ki67 immunostaining performed in 6 out of 12 cases showed a proliferation index not higher than 40%. There was a monotypic expression of IgM in 6 cases (6/12), with monotypic surface immunoglobulin kappa light chain in 5 cases according to blood immuno-globulin light chain.
Skin biopsies of patients in group 2 showed a normal epidermis and a total dermal and hypodermal infiltrate with a nodular or diffuse (2/5), reticular (1/5) or periadnexal (1/5) pattern, mainly composed of large cells, with mature plasma cells noted in only one case. The large cells were positive for CD20 (5/5), BCL2 (5/5) and MUM 1 (2/3), and negative for CD10 (4/5), CD23 (4/5). The Ki67 was generally increased (mean 75%). A monotypic surface immunoglobulin light chain, similar to the isotype of the serum monoclonal component was found in 2 cases (1 kappa and 1 lambda).
MYD88 L265P mutation
MYD88 L265P mutation was tested on the skin biopsy specimens of 6 patients in group 1 and was positive in all cases. In these patients, this mutation was also tested in the blood in 4 cases and was positive. In group 2, only 2 patients were tested for MYD88 L265P mutation: in one patient it was detected by skin analysis and in the other by blood analysis. Overall, all patients (including the 2 patients with initial diagnosis of MZL) tested for MYD88 L265P mutation had a positive result.
Treatment and follow-up data
First- and second-line therapies in both groups after occurrence of cutaneous lesions are shown in Table III.
In group 1, 11/12 patients received a systemic therapy at the time of skin involvement. In 7 of these 11 cases, haematological criteria indicating treatment were present, while in the other 4 cases the only reason to treat was the occurrence of skin lesions. First-line treatments in this group most often consisted of a combination of rituximab and a single chemotherapeutic agent. One patient did not receive any treatment because he had no haematological criteria indicating treatment and did not report skin involvement. One responder patient achieved a complete cutaneous and haematological response after rituximab therapy, while 9 patients had a partial response and one patient had a progressive disease.
In group 2, all patients received treatment at the time of skin involvement and histological transformation. First-line treatment generally consisted of a combination of rituximab and polychemotherapy. One patient received radiotherapy for his skin lesions. A complete response was obtained in 2 cases, one after rituximab-ifosfamid-carboplatine-etoposide (RICE) and one after R-CHOP, followed by recurrence in one case.
Table III. Treatment for Waldenström’s macroglobulinaemia with cutaneous involvement
In group 1, the median follow-up time was 6 years (0.5–21 years) after the diagnosis of WM and 4.5 years (0.5–20 years) after the onset of skin involvement. One patient died from WM. The 3- and 5-year-specific survival rate was 88% (Fig. 2a). In group 2, the median follow-up time was 5.5 years (0.25–10 years) after the diagnosis of WM and 1.4 years (0.25–3 years) after skin involvement. Five patients (71%) died from transformed WM. The 3-year-specific survival rate was 22% (Fig. 2b).
Fig. 2. Specific survival in patients with cutaneous involvement in (a) non-transformed Waldenström’s macroglobulinaemia (WM). (b) WM with histological transformation to diffuse large B-cell lymphoma in the skin.
Extramedullary localization of WM is rare (<5%) and can involve various organs and tissues, including the skin in 5% of cases (9). Histological transformation of WM to DLBCL is also very rare. In contrast with non-transformed WM, transformed WM is associated with extranodal involvement in more than 90% of cases (9), including the skin in 12% (4). Overall, only a few case reports and a few small case series of WM involving the skin have been published (6, 7, 10–15).
This study has some limitations; it is a retrospective study conducted over a long period of time in multiple centres with heterogeneous practices; the method for identifying cases was not homogeneous, including both a dermatological recruitment of mainly non-transformed cases identified by the GFELC, and a haematological recruitment of transformed cases, which prevented us from drawing epidemiological conclusions concerning incidence and frequencies. However, to our knowledge, our study provides the largest series to date of WM involving the skin. It made it possible to characterize and compare the clinical presentation of non-transformed and transformed cases. Because of the long study period, long-term outcome in both groups could be reported.
Some 42% of patients developed skin lesions before the diagnosis of WM, making the dermatological examination the origin of the diagnosis. Despite the polymorphism of skin lesions, we identified a recurrent, homogeneous clinical aspect in a subgroup of 4 patients with non-transformed WM. This easily identifiable clinical picture consisted of non-ulcerated, erythematous-violaceous plaques infiltrating symmetrically and bilaterally the cheekbones, ears and forehead. This clinical aspect has been described previously in 2 case reports. In 1982, Mascaro et al. (10) first reported a patient with diffuse and purplish infiltration of both ears whose skin biopsy specimens showed a dermal infiltration by lymphoplasmocytic tumour cells secondarily linked to WM. Similarly, in 2003, Chan et al. (14) described a woman with prominent violaceous dermal nodules and confluent plaques over the lower forehead, eyebrows, glabella, earlobes and upper cheeks. The skin biopsy showed a lymphoplasmacytic tumour cell infiltrate and the blood and bone marrow analyses were consistent with WM. Although a preferential location on the face, especially a symmetrical infiltration of the ears, has already been described in other haematological B-cell malignancies, such as chronic lymphocytic leukaemia (CLL) (16), extensive, symmetrical violaceous plaques including both the ears and the centre of the face, as observed in our series, seems widely suggestive of cutaneous WM and should be investigated accordingly.
Patients in group 2 showed a different, less specific clinical picture, with nodules and tumours, often rapidly growing and ulcerated, mainly located on the trunk and lower limbs. In the lower limbs, the clinical and pathological presentation could first be suggestive of a primary cutaneous diffuse large B-cell lymphoma, leg type (PCDLBCL-LT).
The histological characteristics of cutaneous lesions of WM have been poorly described and the diagnosis may be difficult on skin biopsies only, as confirmed in our series. The main differential diagnoses are PCDLBCL-LT in transformed cases and MZL in non-transformed cases (17). In our study, skin lesions were first suggestive of primary cutaneous MZL in 3 cases. In one of these patients, first presenting with small skin lesions restricted to the eyelid and the calf, a WM was diagnosed 7 years later on haematological criteria. Skin biopsies were reviewed and further analysed, and the MYD88 L265P mutation was identified, leading to the diagnosis of cutaneous involvement in WM. In the 2 other cases, the diagnosis of primary cutaneous MZL was first considered on pathology reports, but skin involvement in WM was finally diagnosed in the clinical and haematological context (bone marrow biopsy). MYD88 L265P mutation was positive in the skin in one of these 2 cases and was not investigated in the other. In a recent monocentre German study (18), 9 out of 23 patients with a diagnosis of primary cutaneous MZL had a blood paraproteinaemia, corresponding in 4 cases to a monoclonal IgM, and more frequent relapse were observed in these patients than in the group with primary cutaneous MZL and no paraproteinaemia. As bone marrow examination was performed only in selected cases in this series and the MYD88 L265P mutation analysis was not mentioned, it remains possible that some of these patients had undiagnosed WM, suggesting that the frequency of WM with cutaneous involvement could be underestimated.
MYD88 L265P mutation has been identified as a specific blood molecular marker present in 91% of WM patients (19). Conversely, it is infrequent in IgM monoclonal gammopathies of uncertain significance (MGUS, 10–50% of cases) and in MZL (7%), and is completely absent in multiple myeloma. (19–21). Although relatively rare in systemic DLBCL (29%) (22), this mutation has been detected with a high frequency (57%) in PCDLBCL-LT, and was shown to have an independent negative prognostic value in this rare subtype of primary cutaneous B-cell lymphoma (23). The detection of this mutation in skin biopsy specimens of patients with skin involvement in WM has been reported occasionally (7, 24). In the current study, it was present in all cases tested (7/7: 6 in the group of non-transformed cases and one in the other group). Therefore, the presence of MYD88 L265P mutation constitutes an important diagnostic tool for the distinction between primary cutaneous MZL and skin involvement of WM. In cutaneous large B-cell tumours, however, it is less distinguishing, since most PCLBCL-LT express the same mutation as in WM, suggesting that a monoclonal IgM paraprotein should be tested, and an appropriate blood and systemic evaluation should be performed in any case of PCDLBCL-LT.
Asymptomatic patients with WM generally do not require a specific treatment. Criteria for systemic therapy include general symptoms, deleterious IgM activity, cytopaenia or a significant tumour mass (25). In our series, however, 4 patients in the group of non-transformed WM (one with simultaneous skin and haematological diagnosis and 3 with isolated cutaneous progression) received a systemic therapy because of their cutaneous lesions, although they did not meet the classic haematological criteria for treatment. In the group of transformed cases, the skin was the site of histological transformation to DLBCL, justifying in most cases treatment with rituximab and polychemotherapy (generally CHOP), as usually recommended (26).
Extramedullary involvement does not appear to be a poor prognostic factor in non-transformed cases of WM (9). The current study on skin involvement confirms and complements these data, showing a 5-year-survival rate of 88% in patients with cutaneous lesions and no histological transformation, similar to the usual survival of patients with WM. In contrast, histological transformation to DLBCL appears to be a milestone in the course of the disease (4, 27). Indeed, 71% of patients in group 2 died of their disease, resulting in a 3-year specific survival rate of 22%. Whether an early diagnosis of the histological transformation may improve the prognosis remains unknown. However, dermatologists and haematologists should be aware that the skin may be the first site of transformation, and a skin biopsy of any suspicious lesion should be performed in patients with WM, whatever the haematological status.
This study highlights 2 types of cutaneous involvement in WM, with quite different clinical presentation and prognosis. In both types, skin involvement may precede and reveal the haematological disease, leading to an earlier diagnosis. In non-transformed cases, we identified a recurrent clinical picture consisting in erythematous-violaceous plaques infiltrating the cheekbones, ears, and forehead symmetrically and bilaterally. In presence of this clinical aspect, haematological exploration for WM should be performed. In addition, the appearance of cutaneous nodules or tumours during the course of WM should suggest a histological transformation to DLBCL, associated with poor prognosis.
In any case, the diagnosis of such lesions is based on close collaboration between dermatologists and haematologists, allowing analysis of the clinical context, histology, immunohistochemistry and the detection of MYD88 L265P mutation.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize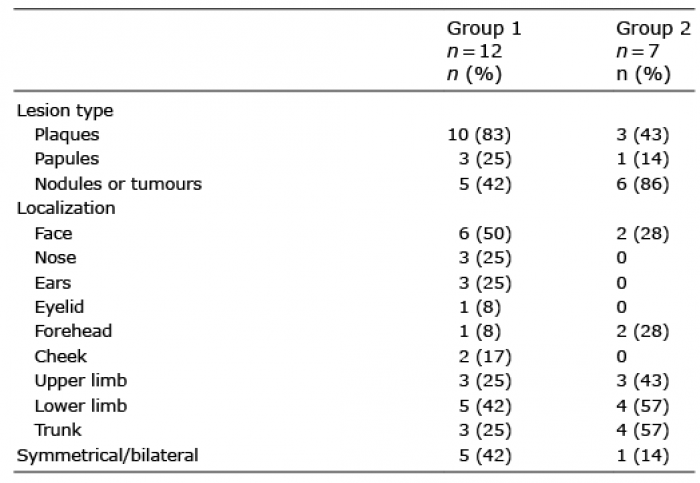 Click to show fullsize
Click to show fullsize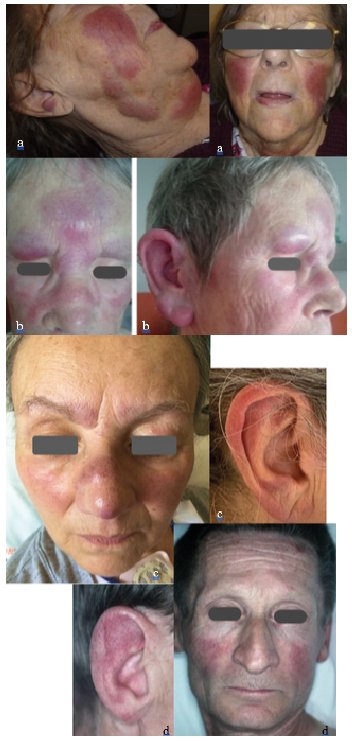 Click to show fullsize
Click to show fullsize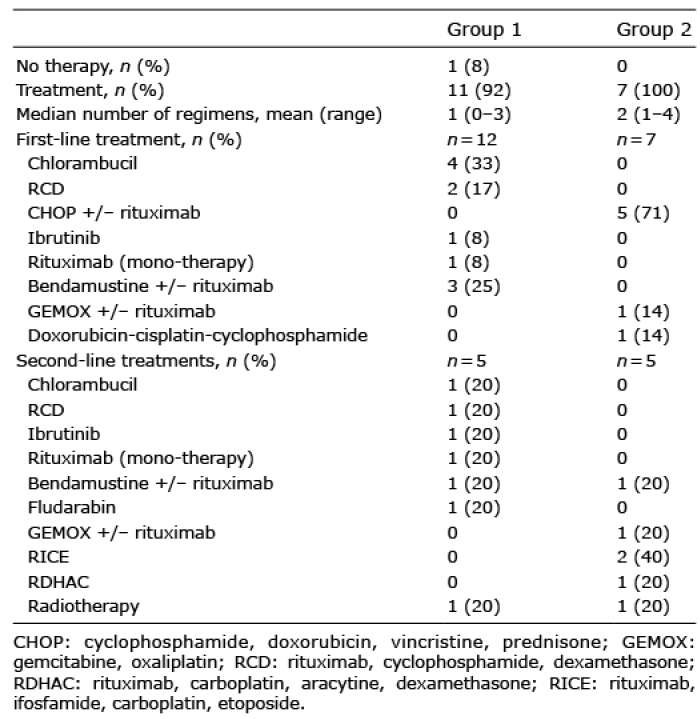 Click to show fullsize
Click to show fullsize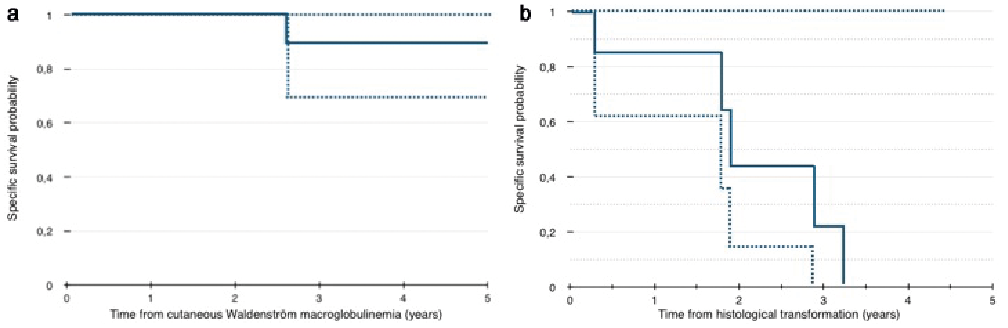 Click to show fullsize
Click to show fullsize