


1Department of Dermatology, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, 2Institute of Clinical Medicine, College of Medicine, National Cheng Kung University, 3Center of Clinical Medicine, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, 4School of Medicine, College of Medicine, National Cheng Kung University, Tainan, Taiwan, 5St John’s Institute of Dermatology, King’s College London (Guy’s Campus), London, UK, 6Department of Dermatology, Hokkaido University Graduate School of Medicine, Sapporo, Japan, and 7International Center for Wound Repair and Regeneration (iWRR), National Cheng Kung University, Tainan, Taiwan. E-mail: natsuga@med.hokudai.ac.jp; kylehsu@mail.ncku.edu.tw
Accepted Jul 7, 2020; Epub ahead of print Jul 29, 2020
Acta Derm Venereol 2020; 100: adv00242.
Plectin is a linker-protein that is expressed ubiquitously in many tissues, including skin, muscle and nervous system. Plectin interacts with intermediate filaments and hemidesmosomal proteins, including the β4 integrin subunit (1), and binding of plectin to α6β4 integrin is the critical step in hemidesmosome assembly (2). Mutations in the plectin gene (PLEC) can underlie both autosomal dominant (AD) and autosomal recessive (AR) subtypes of the inherited blistering disease, epidermolysis bullosa simplex (EBS), the latter may also include extracutaneous features of muscular dystrophy or pyloric atresia (1). Typically, mutations in PLEC causing AR-EBS are nonsense mutations or out-of-frame indels. In contrast, missense mutations in PLEC have been reported in autosomal dominant (AD)-EBS, but rarely identified in AR-EBS (3). We present here 2 new cases of AR-EBS caused by compound heterozygous mutations in PLEC, in which one of the mutations in both subjects was atypical: a novel missense mutation. Moreover, we demonstrate the pathogenicity of the missense mutation by in vitro transfection studies.
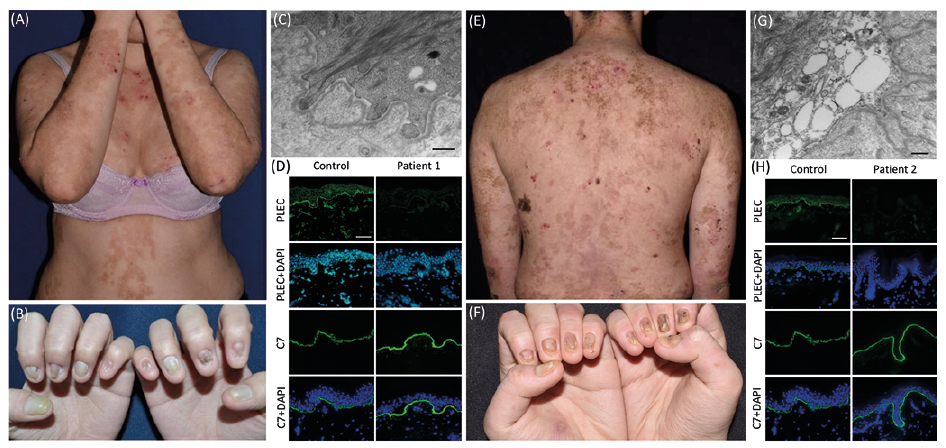
Case 1. A 31-year-old woman presented with painful generalized blistering and erosions since birth. Physical examination showed multiple erythematous blisters and erosions as well as hyperpigmented patches on the trunk and extremities, dystrophy of all 20 nails, a few small erosions in the oral cavity, and multiple hyperkeratotic plaques on the soles (Fig. 1A, B). She had no clinical muscle weakness, hoarseness, ocular lesions, or other extracutaneous manifestations. Neither of her parents, age 58 and 60 years, had any blisters, erosions, or nail abnormalities. Transmission electron microscopy (TEM) study of a skin biopsy revealed hypoplastic hemidesmosomes and focal reduplication of the lamina densa (Fig. 1C). Plectin labelling was greatly diminished at the dermo-epidermal junction (Fig. 1D). Whole exome sequencing (WES) evaluating all 21 genes implicated in EB found 2 heterozygous PLEC mutations (according to NM_000445.5, transcripts for plectin 1c isoform), c.6955C>T (p.Arg2319Ter) in exon 31 reported in (4), and previously unreported c.956T>C (p.Leu319Pro) in exon 9. In silico analyses predicted both mutations to be deleterious (p.Arg2319Ter, CADD: 36. p.Leu319Pro, CADD: 24). Sanger sequencing confirmed the segregation of these 2 PLEC mutations in the family (Fig. S1A).
Fig. 1. Clinical manifestations and the results of immunofluorescence (IF) and transmission electron microscopy (TEM). (A) The 31-year-old woman (Case 1) has generalized blistering and nail dystrophy; (B) In Case 1, TEM shows hypoplastic hemidesmosomes and irregular reduplication of basement membrane beneath the basal keratinocytes. Scale bar=500 nm (C) In Case 1, IF staining of plectin is significantly reduced, while type VII collagen (C7) expression is normal. Scale bar=50 µm; (D) The 18-year-old man (Case 2) has generalized blistering and nail dystrophy (E) In Case 2, TEM shows vesicles in the basal keratinocytes and hypoplastic hemidesmosomes. Scale bar=500 nm; (F) IF staining of plectin is almost completely lost, in contrast to normal C7 expression. Scale bar=50 µm.
Case 2. An 18-year-old man presented generalized erosions and blisters since birth. Physical examination revealed similar diffuse erythematous blistering and erosions, as well as hyperpigmented patches on the trunk and extremities. In addition, he had multiple small blisters in the oral cavity, palmoplantar keratoderma, and dystrophy of all 20 nails (Fig. 1E, F). He had no muscle weakness, hoarseness, ocular lesions, or other extracutaneous manifestations. Neither of his parents, age 43 years and the other of unknown age, had any blisters, erosions, or nail abnormalities. TEM of a skin biopsy revealed vesicles in basal cells accompanied by hypoplastic hemidesmosomes (Fig. 1G). Immunofluorescence studies showed near-complete absence of plectin at the dermo-epidermal junction (Fig. 1H). WES of case 2 found 2 PLEC mutations, c.956T>C (p.Leu319Pro) and a previously unreported c.2807G>A (p.Trp936Ter) in exon 22; in silico analyses predicted both mutations to be disease-associated (p.Trp936Ter: CADD: 40). Sanger sequencing also revealed c.2807G>A in his father (Fig. S1B). The presence of c.956T>C could not be confirmed in her mother, due to lack of a maternal DNA sample.
In contrast to the truncation mutations, p.Leu319Pro is not expected to affect PLEC gene expression. It was hypo-thesized that p.Leu319Pro of plectin has an impact on protein stability or protein-protein interaction. To test this hypothesis, 3 V5-tagged vectors were prepared with PLEC cDNA fragments spanning its actin-binding domain, which contains Leu at position of 319 and is a β4 integrin-binding site (5): PLEC1a_1-315 wild type, p.Leu319Pro (in which p.Leu292Pro of plectin 1a, equivalent to p.Leu319Pro of plectin 1c, was introduced), and p.Leu319dup (in which p.Leu292dup of plectin 1a, a previously reported mutation (6), was introduced). Plectin 1a rather than other isoforms was utilized because this isoform is preferentially expressed in the hemidesmosomes of epidermal keratinocytes (7). The 3 types of PLEC constructs were transfected into HEK293 cells. The lysates of the cells transfected with these constructs alone showed similar bands by immunoblotting (Fig. S1C). However, when the ITGB4 cDNA construct (FLAG-tagged vector) spanning a.a.1115-1355, which is a plectin binding site (8), was cotransfected into HEK293 cells, the amount of plectin and β4 integrin was greatly diminished in p.Leu319Pro or p.Leu319dup-transfected cells (Fig. S1C) (see Appendix S1). These data indicate that mutations at Leu319 in plectin lead to protein instability, especially when β4 integrin is present with plectin.
PLEC mutations may cause EBS-AD or EBS-AR. However, c.956T>C (p.Leu319Pro) is an unusual pathogenic mutation for EBS-AR, since most PLEC mutations underlying EBS-AR are nonsense, frameshift, or splice site mutations (1). Some missense or in-frame del/ins mutations in PLEC have been reported in EBS-AR (1, 4, 9), but the pathogenicity of the mutations have rarely been examined except for a few examples: p.Leu319dup to cause self-aggregation of plectin as well as impaired plectin-β4 integrin binding (6) and p.Phe755del to hinder plectin-COL17 binding (10). Our in vitro assay clearly showed that p.Leu319Pro or p.Leu319dup reduced plectin and β4 integrin proteins in the cells, which were cotransfected with ITGB4 constructs, implying that the misassembled complex of plectin and β4 integrin by the mutations enhances protein degradation and leads to defective hemidesmosome formation. Phenotypically, however, p.Leu319Pro does not appear to be associated with extracutaneous abnormalities, since recessive loss-of-function mutations in exon 31 (as in Case 1) can be associated with muscular dystrophy, and other recessive loss-of function mutations outside this exon (as in Case 2) can result in pyloric atresia (1). In both our patients, the clinical abnormalities were limited to the skin. However, the late-onset muscular dystrophy cannot be excluded.
In conclusion, these 2 cases shared a novel recessive missense mutation in PLEC, p.Leu319Pro, which is similar to the site of a previously reported mutation, p.Leu319dup, highlighting the importance of this location in the proper assembly of plectin and β4 integrin.
This work was financially supported by the International Center for Wound Repair and Regeneration at National Cheng Kung University from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan, and by the grants from Health Promotion Administration, Ministry of Health and Welfare (rare disease prevention and control work and research projects) in Taiwan.


 Click to show fullsize
Click to show fullsize