


1Department of Dermatology, University Hospital of Munich (LMU), Frauenlobstrasse 9–11, DE-80337, Munich, 2Clinic of Pediatric Hematology and Oncology, University Medical Centre, Hamburg-Eppendorf, Hamburg, and 3Department of Pathology, Hematopathology Section and Lymph Node Registry, University Hospital Schleswig-Holstein, Kiel, Germany. E-mail: Kinan.hayani@med.uni-muenchen.de
Accepted Jul 9, 2020; Epub ahead of print Jul 29, 2020
Acta Derm Venereol 2020; 100: adv00245.
Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is primarly a disease of the elderlies with an overall infaust prognosis. Cases of pediatric BPDCN are only rarely published. Here we present a case of a juvenile BPDCN with a currently stable disease course.
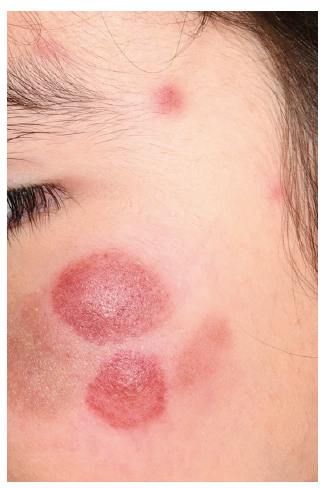
An 8-year-old girl presented with asymptomatic, red-brown, haemorrhagic nodules and plaques on her cheeks, trunk and lower extremities (Fig. 1). She was otherwise in good health with no systemic symptoms and an inconspicuous medical history. According to her parents, these nodules had progressively increased in size and number over the last 9 months. Preliminary serological workup was normal.
Fig. 1. Asymptomatic, red-brown, haemorrhagic plaques and nodules of different sizes on the face of an 8-year-old girl.
A skin biopsy revealed a nodular dermal and subcutaneous infiltration of large atypical blastoid cells with a CD4+CD56+CD123+CD20–CD34–CD79a–CD163–Myeloperoxidase–lysozyme– immunophenotype (Fig. 2). Bone-marrow biopsy revealed infiltration (2%) with the same blastic cells. No involvement of peripheral blood, lymph nodes, neuromeningeal tissues or other organs was detected.
Fig. 2. Histopathology of blastic plasmacytoid dendritic cell neoplasm in (a) skin and in (b) bone marrow, haematoxylin and eosin staining. The neoplastic cells show immunoreactivity to (c, d) CD4, (e, f) CD56 and (g, h) CD123 and (i, j) negativity for myeloperoxidase.
Based on the clinico-pathological correlation the diagnosis of blastic plasmacytoid dendritic cell neoplasm (BPDCN) was retained. The patient was then referred to a paediatric haemato-oncological clinic for further investigation and treatment. She received a polychemotherapy (acute lymphoblastic leukaemia (ALL) high-risk treatment-protocol) consisting of 12 different cytostatic agents given sequentially: Adriamycin/daunorubicin, asparaginase (Aspa), cytarabine, cyclophosphamide, dexamethasone, etoposide, methylprednisolone, 6-mercaptopurine, methotrexate (MTX), thioguanine and vincristine. This therapy was well-tolerated and the patient is currently in complete remission. With this very good remission achieved, a maintenance therapy will be continued until 2 years after diagnosis. Allogenic haematopoietic stem cell transplantation (allo-HSCT) is currently not considered.
BPDCN is an exceedingly rare and aggressive tumour, derived from precursors of plasmacytoid dendritic cells, usually affecting elderly males. To date, only a few cases of paediatric BPDCN have been reported. Currently BPDCN is considered as a distinct entity in the WHO classification of tumours of hematopoietic and lymphoid tissues (1). The skin is usually the site of presentation of the disease; however, BPDCN can also be systemic without skin involvement.
The aetiopathogenesis of the disease is still unclear; however, associations with other myelodysplastic syndromes as well as genetic aberrations in cell-cycle checkpoints have been reported (2). No associations with oncogenic viruses (e.g. EBV, HTLV-1) have been found.
The definitive diagnosis of BPDCN is made histologically. Several immunohistochemical markers are helpful for the diagnosis of BPDCN. In most reported cases, the following markers, amongst others, were positive in the blastic cells of the tumour: CD4, CD56, CD123, CD303, TCL-1, TdT and ICSBP/IRF8. In contrast, CD3, myeloperoxidase, granzyme-B and lysozyme are almost always non-reactive (3).
The biological behaviour of BPDCN is usually aggressive, often with a fatal outcome within months (4). This contrasts with the initial, often innocuous, clinical presentation with merely cutaneous involvement, as in the current patient. Interestingly however, the prognosis of BPDCN is assumed to be better in paediatric patients. An immediate haemato-oncological referral and multidisciplinary management is warranted. Due to the rarity of the disease, no standardized therapy exists currently, most of the studies are of retrospective nature, and the vast majority of patients included are elderly adults. Treatment with high-risk ALL-type, as well as AML-type chemotherapy regimens, appears to be effective. Aspa-MTX containing regimens achieve the best response rate with a lower toxicity profile, possibly paving the way for HSCT and eventual long-term complete remission of the disease (5). However, another report suggests that a good initial response to polychemotherapy will often be followed by therapeutic resistance (6). Aberrations detected in BPDCN in the NF-kappaB pathway may serve as therapeutic target in the future (7). Recently, Tagraxofusp, a CD123-directed cytotoxin consisting of recombinant human interleukin-3 fused to a truncated diphtheria toxin, showed promise, in spite of reported serious side-effects (e.g. capillary leak syndrome); yet, studies in paediatric BPDCN are lacking (8). Allo-HSCT can be reserved for children who relapse and have subsequently achieved a second complete remission with chemotherapy (6). In conclusion, prospective trials to standardize the therapeutic ladder in juvenile BPDCN are lacking and should be encouraged.


 Click to show fullsize
Click to show fullsize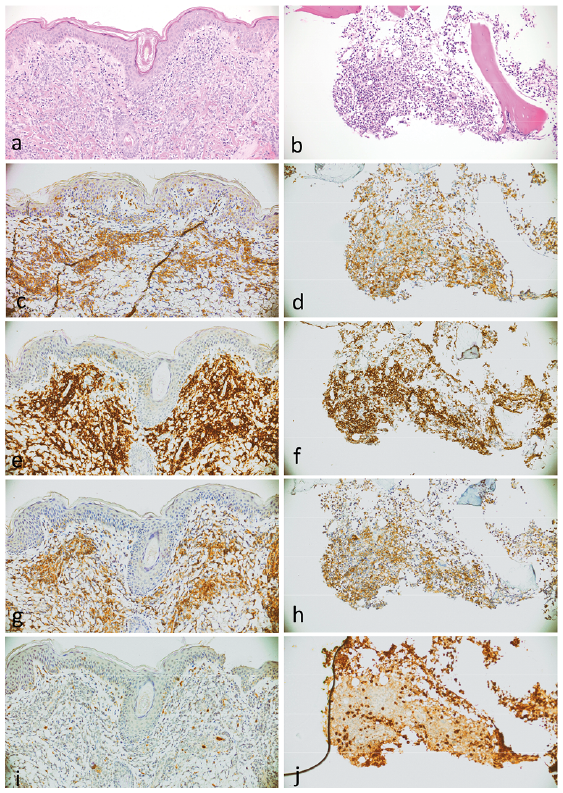 Click to show fullsize
Click to show fullsize