


1Division of Cutaneous Science, Department of Dermatology, Nihon University School of Medicine, 30-1 Oyaguchi-Kamicho, Itabashi-ku, Tokyo 173-8610, and 2Department of Anatomy, Nihon University School of Dentistry, Tokyo, Japan. *E-mail: hayama-nhn@umin.ac.jp
Accepted Sep 7, 2020; Epub ahead of print Sep 14, 2020
Hidradenitis suppurativa (HS) is a chronic, inflammatory, recurrent, debilitating disease of hair follicles with painful, deep-seated, inflamed lesions. Symptoms typically appear in the apocrine gland-bearing areas of the body, especially the axillae, and inguinal and anogenital regions. It has a risk of developing cutaneous squamous cell carcinoma (SCC) (1). In Western countries, some patients with HS have a family history of this disease, and an autosomal dominant trait has been reported in one-third of familial HS (2). Recently, loss-of-function mutations in the presenilin 1 (PSEN1), presenilin enhancer 2 (PSENEN), and nicastrin (NCSTN) genes, encoding key components of the γ-secretase complex, have been identified as a cause of familial HS in Chinese, Japanese, and French families (3). Mutations in genes coding γ-secretase subunits seem to be responsible for approximately 5% of HS cases (1).
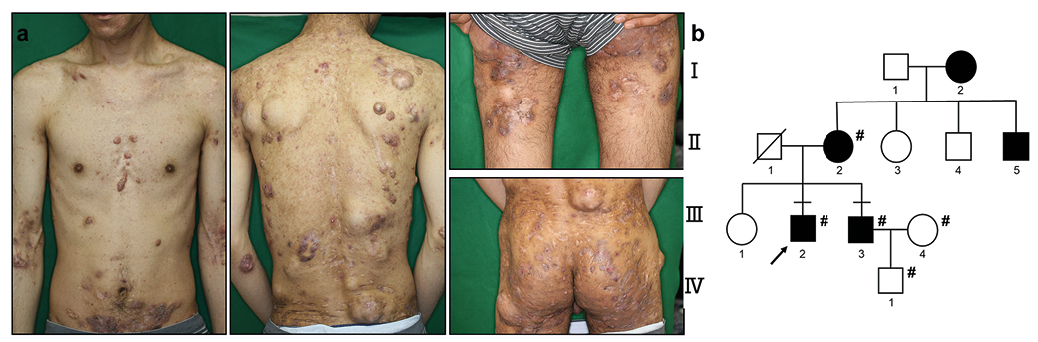
A 36-year-old man presented to our outpatient clinic with a 21-year history of relapsing nodules on his face, body, and limbs and abscesses on his buttocks. His mother, younger brother, maternal grandmother, and an uncle also had similar symptoms. No diabetes mellitus, hirsutism, inflammatory bowel diseases, or obesity were observed in this family. Only his grandmother was a smoker.
At initial presentation, various-sized multiple nodules and pustules were distributed on the body and limbs (Fig. 1a). He underwent multiple surgeries and took a range of antibiotics. These treatments were not sufficiently effective, but palliative. Six years after the patient’s first visit, a tumour developed on his right buttock, and a diagnosis of SCC was made based on histopathological findings. Despite the treatment with chemotherapy, radiotherapy, and surgical excision, the tumour relapsed and metastasized to the sacral bone and inguinal lymph nodes. He died of SCC 8 years after the initial visit.
In this family, 5 members were affected and 7 were unaffected by HS. Five of the 12 individuals were recruited for gene analysis. Among them, the proband (III-2) and his younger brother (III-3) presented to us and were diagnosed with HS (Fig. 1b), and the brother was treated with antibiotics. According to a medical interview, proband’s maternal grandmother (I-2) and mother (II-2) had a milder HS phenotype (Fig. 1b), but their treatments were unknown.
The participants or their legal guardians provided written informed consent in compliance with the principles of the Declaration of Helsinki. The study was approved by the Medical Ethics Committee of Nihon University (permission number: 194-0). HS was clinically diagnosed based on the European S1 guideline (1). Blood and saliva samples were obtained from 3 affected and 2 unaffected members of the family. Genomic DNA was extracted, and PCR amplification of PSEN1, PSENEN and NCSTN genes was performed as described in Appendix S1 and Table SI.
A heterozygous single-nucleotide mutation c.1285C>T (p. R429X) in the exon 11 of NCSTN gene (reference sequence GenBank accession number NM_015331.2) was detected in the proband (III-2), his younger brother (III-3), and his mother (II-2) (Fig. S1). Notably, the unaffected 2 individuals showed wild-type sequence (III-4 and IV-1). By reference to gnomAD v2.1.1, we have confirmed that this sequence variant has not been reported.
To determine whether the NCSTN mutation identified in this family affects nicastrin structure, we assessed amino acid change at the mutation site. The mutation was found to be a nonsense mutation, leading to the truncation of the nicastrin protein at codon 429. This mutation is located on the extracellular domain of nicastrin.
Fig. 1. Clinical features of the proband and the pedigree of the familial hidradenitis suppurativa. (a) The proband presented severe clinical features. (b) The family tree indicates an autosomal dominant inheritance. Solid symbols denote affected individuals and open symbols denote unaffected individuals. #Denotes individuals who were examined for gene mutation.
Japanese HS is characterized by male predominance and less family history compared with Western cases (4). There are 2 reports of familial HS with genetic mutations in Japan (5, 6). Nomura et al. (5) have reported Japanese familial HS, where 4 out of 12 males (33.3%) and one out of 17 females (5.8%) had HS. Furthermore, in the report, the proband’s sister, a 70-year-old carrier having the same mutation, presented no HS. Interestingly, in our case, a genetic mutation was also found in the mother and maternal grandmother, but their clinical symptoms were milder than those of males.
Nicastrin, encoded by NCSTN gene, is a critical subunit of the γ-secretase complex (7). It is involved in the integration of different subunits into the functional γ-secretase complex and in complex stabilization. This enzyme complex cleaves the intracellular domain of Notch (8). In the skin, Notch signalling plays a pivotal role in hair follicle differentiation and epidermal barrier function maintenance. It also acts as an epidermal tumour suppressor (8). Most mutations found in genes encoding γ-secretase subunits in HS are loss-of-function variants, leading to reduced protease activity and attenuated Notch signalling (8). In particular, haploinsufficiency of the NCSTN gene caused by the nonsense mutation has shown to contribute to the occurrence of familial HS (9). Disruption of the Notch signalling pathway causes follicular keratinization, follicular atrophy, epidermal cyst formation, and epidermal hyperplasia (3). The current study found the novel NCSTN gene nonsense mutation, exon 11: c.1285C>T, which results in truncated form of nicastrin protein. Mutations that result in premature termination codons in exon 5, 11, and 15 of NCSTN gene have been shown to be responsible for familial HS (9, 10). Indeed, all 3 symptomatic members in our patient’s family had the heterozygous c.1285C>T nonsense mutation in exon 11, and neither of the unaffected 2 individuals did. Therefore, based on the criteria proposed by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (11), we considered this DNA variant as pathogenic. In addition, the occurrence of HS in the individuals with heterozygous mutation indicate the possibility of haploinsufficiency of NCSTN gene, as with the previously reported cases carrying heterozygous nonsense mutation in exon 11 of the same gene (9).
HS patients have a risk of SCC, with an overall incidence of 1–3.2% (12), but its pathogenesis is unclear. The mean latency period from diagnosis of HS to the development of SCC is approximately 27 years (13). Patients with HS who developed SCC in HS-affected sites revealed a remarkable male dominance (86%), and 57% of the patients died within 2 years after the diagnosis of SCC (12). Our proband patient developed SCC 27 years after the onset of HS and died 2 years after the diagnosis of SCC. The loss-of-function mutation in our patient might be involved in the development of SCC, because Notch acts as an epidermal tumour suppressor in SCC (8).
In conclusion, a novel heterozygous NCSTN mutation c.1285C>T (p. R429X) was found in a Japanese familial HS. Clinical symptoms largely varied among the individuals even though the same genetic mutation was shared, indicating that other factors may also affect the severity of the disease.
The authors thank the patients and their families for participating in this study. We also thank Ms Asako Oguni for her technical assistance.
Conflicts of interest: HK and FH received honoraria for speaker and consultancy from AbbVie and Eisai. TT received honoraria for speaker and consultancy from AbbVie.


 Click to show fullsize
Click to show fullsize