


1Division of Allergy and Immunology, and 2Department of Dermatology, Venereology and Allergology, Charité – Universitätsmedizin Berlin, Berlin, Germany
Intravenous immunoglobulins are an effective and well-tolerated treatment option for immune dermatological diseases. However, they are primarily used to treat diseases with a severe course and are mostly used off-label. Therefore, it is important to document case series on the use of intravenous immunoglobulins in rare immune dermatological diseases. We present here 32 patients who were treated with intravenous immunoglobulins in our clinic between 2015 and 2020. The indications were dermatomyositis, including amyopathic and paraneoplastic forms, as well as overlap- syndromes (n = 18), pemphigus vulgaris (n = 2), mucous membrane pemphigoid (n = 2), linear IgA dermatosis (n = 1), necrotizing vasculitis (n = 1), urticarial vasculitis (n = 1), systemic scleroderma (n = 1), Stevens-Johnson syndrome (n = 1), pyoderma gangrenosum (n = 3), and livedoid vasculopathy (n = 2). The data from this case series confirm the efficacy and tolerability of intravenous immunoglobulins as an adjuvant treatment option for rare therapy-resistant immune dermatoses.
Key words: intravenous immunoglobulin; dermatomyositis; autoimmune bullous dermatosis; vasculitis; Stevens-Johnson syndrome; pyoderma gangrenosum.
Accepted Oct 7, 2020; Epub ahead of print Oct 13, 2020
Acta Derm Venereol 2020; 100: adv00298.
doi: 10.2340/00015555-3662
Corr: Margitta Worm, Division of Allergy and Immunology, Department of Dermatology, Venereology and Allergology, Charité – Universitätsmedizin Berlin, Charitéplatz 1, DE-10117 Berlin, Germany. E-mail: margitta.worm@charite.de
Intravenous immunoglobulins are an effective and well-tolerated treatment option for immune dermatological diseases. However, they are primarily used to treat diseases with a severe course and are mostly used off-label. Therefore, it is important to document case series on the use of intravenous immunoglobulins in rare immune dermatological diseases. We present here 32 patients with various immune dermatological indications who were treated with intravenous immunoglobulins in our clinic between 2015 and 2020. The data from this case series confirm the efficacy and tolerability of immunoglobulins as an adjuvant treatment option for rare therapy-resistant immune dermatoses
Intravenous immunoglobulins (IVIg) are used successfully in dermatology for the treatment of severe forms of autoimmune diseases and toxic epidermal necrolysis (1). Numerous case series and case reports also suggest their successful use in a large number of other rare immune dermatological diseases. However, to date, there have only been a few randomized controlled trials (RCT) (2–6). One of the limitations in the use of IVIg is the high costs of therapy, which require careful indication, mostly for the treatment of severe courses of rare diseases (7). Although IVIg are therefore used very selectively, and larger case series are not available or are available only to a limited extent, they often offer an excellent therapeutic option due to their broad spectrum of action and good tolerability.
The aim of this study was to describe the course of therapy in a case series of patients from our clinic who were treated with IVIg between 2015 and 2020. For this purpose, the study specifically examined the response to and duration of the therapy, as well as the side-effects that were documented in the various immune dermatological indications, including dermatomyositis, vasculitis, Stevens-Johnson syndrome, and autoimmune bullous dermatoses.
A total of 32 patients who were treated with IVIg in our clinic between January 2015 and March 2020 were studied retrospectively. The cohort includes 24 female and 8 male patients, with a median age of 71.5 years (range 28–92 years) at the first administration of IVIg. The indications were dermatomyositis, including amyopathic and paraneoplastic forms, as well as overlap-syndromes (n = 18), pemphigus vulgaris (n = 2), mucous membrane pemphigoid (n = 2), linear IgA dermatosis (n = 1), necrotizing vasculitis (n = 1), urticarial vasculitis (n = 1), systemic scleroderma (n = 1), Stevens-Johnson syndrome (n = 1), pyoderma gangrenosum (n = 3), and livedoid vasculopathy (n = 2).
The main clinical characteristics of the patient cohort are shown in Tables I and II. The median time between the initial diagnosis and the first cycle of IVIg was 4.5 years, with an applied dosage of 2 g of IVIg per kg of body weight per cycle in 23 patients. Four patients received 1 g/kg, 4 other patients 0.5 g/kg, and one patient 0.4 g/kg. Dose reduction depended mostly on the patient’s medical history (e.g. history of vein thrombosis or carcinoma) and observed side-effects (i.e. if the reduced dosage was better tolerated). The number of cycles was highly variable, depending on the disease (1–51 cycles). Most repeated cycles are herewith associated with the longest therapy time that was mainly observed in patients with dermatomyositis. The main findings of this study are summarized in Table III and are described below.
Table I. Characterization of the patient cohort (n = 32)
Table II. Dosages and side-effects of intravenous immunoglobulins (IVIg) for the different diagnoses (n = 32)
Table III. Key findings
Dermatomyositis and overlap syndromes
Eighteen patients in the study cohort (15 females and 3 males) had a diagnosis of dermatomyositis. Their median age at first administration of IVIg was 73 years (range 43–92 years). Two patients had an amyopathic and one patient a paraneoplastic dermatomyositis. Seven patients had overlap syndromes, 5 of whom had lupus erythematosus and 2 had systemic scleroderma. IVIg were used mainly as a second- or third-line therapy. After initial treatment with corticosteroids alone or in combination with other immunosuppressants, the patients did not achieve disease control or experienced side-effects. The median time between initial diagnosis and the first administration of IVIg was 2.5 years (range 0–15 years). Co-medications included prednisolone (5–10 mg/day) in 17 patients, as well as (additionally) methotrexate (7.5–15 mg/week), azathioprine (100–150 mg/day), hydroxychloroquine (200–400 mg/day) and/or methylprednisolone (375–750 mg over 3 days every 4 weeks) in 11 patients. The median time between first and last cycle of IVIg during the observation period was one year (range 0–5 years) and the median number of IVIg cycles was 6 (range 1–51 cycles). The median IVIg dosage was 2 g/kg, mostly administered over 2 days, at a median body weight of 60 kg (range 40–100 kg). Three patients received 1 g/kg, and 3 received 0.5 g/kg IVIg. Overall, the therapy was well tolerated. Documented side-effects included diarrhoea, nausea, vomiting, headache and/or abdominal pain in 3 patients. In 17 patients, the therapy resulted in a subjective and objective improvement in symptoms. One patient discontinued therapy after the first administration of 1 g/kg IVIg, due to headache, abdominal pain and lack of efficacy. Two female patients of the cohort received ≥ 6 doses of 1 g/kg IVIg due to a history of venous thrombosis in both legs and side-effects. Despite taking half the dose, improved muscle strength and healing of existing skin lesions were observed in both patients. One of these patients reported a relapse during a follow-up 4 months after the last cycle of IVIg. She received a total of 6 doses; the therapy could not be continued due to the refusal of coverage by the health insurance. As co-medication, she received prednisolone (7.5 mg/day) and methotrexate (15 mg/week). Another female patient received 6 cycles of 0.5 g/kg IVIg in combination with prednisolone (10 mg/day). Despite taking an even lower dosage due to a history of breast cancer, this patient also showed improved muscle strength and skin lesions. She did not relapse until 11 months after the last cycle.
Autoimmune bullous dermatoses
Five patients, 3 female and 2 male patients, received IVIg for the treatment of blistering autoimmune diseases. The diagnoses were pemphigus vulgaris (n = 2), mucous membrane pemphigoid (n = 2), and linear IgA dermatosis (n = 1). The median age at first IVIg administration was 72 years (range 64–84 years). This group of patients also showed a therapy-resistant disease course. The median time between diagnosis and first cycle of IVIg was 5 years (range 1–9 years) and the patients had previously been treated with different immunosuppressants. They received a median of 6 doses of IVIg (range 3–13 doses) over 2–5 days at a dosage of 2 g/kg in 3 patients, and 0.5 and 0.4 g/kg in the 2 other patients at a median body weight of 85 kg (range 70–100 kg). The median time between first and last IVIg cycle was one year (range 0–2 years). Four patients received prednisolone (5–10 mg/day) as co-medication. One patient with pemphigus vulgaris received additional rituximab at a reduced dosage (2 cycles of 500 mg 2 weeks apart) and azathioprine (100 mg/day). Another patient with mucous membrane pemphigoid received methotrexate (10 mg/week). Therapy with IVIg was effective in all 5 patients; 4 of them tolerated the therapy well. One of the patients with relapsing pemphigus vulgaris showed a good response after 6 months of off-label therapy with 0.5 g/kg IVIg despite low dosing. As co-medication, she received prednisolone 10 mg/day, which was reduced to 5 mg/day and then discontinued. However, the patient reported unclear headache and chest pain during IVIg therapy, so she did not wish to continue. In the follow-up, a stable skin condition for at least 8 months was documented, even without therapy. One of the patients with mucous membrane pemphigoid reported severely impaired quality of life. In particular, the oral mucosa and oesophagus were affected and, in the course of the disease, synechiae formed on the inner corner of the left eye and she reported a burning sensation in both eyes. She received an off-label therapy with only 3 cycles of IVIg at standard dosage during the observation period, so that the response could not be ultimately evaluated. Nevertheless, after 3 months of well-tolerated therapy, the disease activity ceased.
Vasculitis
A 51-year-old male patient with many years of existing necrotizing vasculitis of the right forefoot of unknown origin with progressive ulceration and imminent amputation as well as massive pain exacerbation was given IVIg at a dosage of 2 g/kg over 2 days in combination with prednisolone (10 mg/day), pentoxifylline (1,200 mg/day), methylprednisolone (750 mg over 3 days every 4 weeks) and iloprost (Ilomedine) (200 µg over 5 days every 4 weeks). Previous therapies included cyclophosphamide/dexamethasone (16 cycles of 500/300 mg over 3 days every 4 weeks, followed by 3 cycles of dexamethasone), cyclophosphamide (50 mg/day for 5 years), azathioprine (100 mg/day for 5 years) and methotrexate (10 mg/week for 6 months). The IVIg therapy was well tolerated by the patient and he reported improvement. However, no more IVIg doses were administered during the observation period, due to a rejection of reimbursement by the health insurance.
A 76-year-old male patient with therapy-resistant urticarial vasculitis diagnosed 6 years earlier received 2 doses of 2 g/kg IVIg over 2 days 4 weeks apart. The patient reported headaches and an exacerbation of the urticarial symptoms after the first cycle.
Systemic scleroderma
A female patient received 8 cycles of 2 g/kg IVIg over 2–5 days for the treatment of progressive systemic scleroderma with lung and kidney involvement diagnosed 6 years earlier (off-label). Previous therapies included cyclophosphamide/dexamethasone (10 cycles of 500/300 mg over 3 days every 4 weeks and 2 cycles of dexamethasone), cyclophosphamide (50 mg/day for 7 months), and methylprednisolone (3 cycles of 750 mg over 3 days every 4 weeks). No side-effects of the IVIg therapy were documented. The first 6 IVIg doses were given every 4 weeks, and the patient also continued therapy with methotrexate (10 mg/week), which she had started more than 3 years earlier. After the first 6 cycles, the skin condition improved. Approximately 9 months later, the patient received a further 2 doses of IVIg in combination with azathioprine (50 mg/day) when the disease progressed. The patient was then given further rheumatological care.
Stevens-Johnson syndrome
A 28-year-old male patient with Stevens-Johnson syndrome received acyclovir (1500 mg/day for 7 days), ampicillin/sulbactam (6/3 g/day for 9 days), and prednisolone (250 mg/day declining dose). Because of pronounced symptoms, IVIg were additionally administered at a dosage of 2 g/kg over 4 days and were well tolerated. During the therapy, there was a clear improvement in the symptoms. A follow-up was carried out 4 weeks after discharge and showed unremarkable skin findings, except for hyperpigmentation on the back.
Pyoderma gangrenosum
Three female patients received IVIg for the treatment of pyoderma gangrenosum (off-label). The first patient was 82 years old when she received a total of 4 doses of 2 g/kg IVIg over 2 days at 4-week intervals. The therapy was well tolerated and there was a slight improvement in the skin condition. Co-medication included methylprednisolone (750 mg over 3 days every 4 weeks) and azathioprine (100 mg/day). The patient had been diagnosed with pyoderma gangrenosum one year earlier, after 3 years of recurrent impaired wound healing. Previous therapies included prednisolone and antibiotics. Following IVIg therapy, the patient received Infliximab (5 mg/kg at intervals of 2, 4, then every 8 weeks) and the skin lesions healed further.
A second patient received the first IVIg dose at the age of 67 years. She was diagnosed with pyoderma gangrenosum 6 months earlier, and previous therapies included methylprednisolone (3 cycles of 375–750 mg over 3 days every 4 weeks), methotrexate (10–25 mg/week for 4 months) and adalimumab (2 doses of 80 mg one week apart, then 40 mg every 2 weeks for a total of 2 months). Overall, the patient received 11 doses of 2 g/kg IVIg over 2 days every 4 weeks. Co-medication included prednisolone (starting at 50 mg/day and reduced to 20 mg/day) and azathioprine (100 mg/day). The skin condition stabilized after 6 IVIg cycles. The infusions were all well tolerated.
Another 39-year-old obese patient received a single dose of 2 g/kg IVIg over 2 days in combination with methylprednisolone (750 mg over 3 days every 4 weeks). In addition, the patient received ampicillin/sulbactam (6/3 g/day) and prednisolone (120 mg/day declining dose). With the simultaneous occurrence of hidradenitis suppurativa, a PASH syndrome had been diagnosed and the patient had previously been treated with adalimumab (160 mg, then 80 and 40 mg every 2 weeks, then 40 mg/week for a total of 3 months). After one, well-tolerated cycle of IVIg, there was no clear response. At the same time, Crohn’s disease was also diagnosed, and the patient was further treated with ustekinumab (90 mg every 8 weeks for 4 months), mycophenolate mofetil (2,000 mg/day for 4 months), methotrexate (15–20 mg/week) and infliximab (600 mg every 4, then every 8 weeks).
Livedoid vasculopathy
A female patient with livedoid vasculopathy was treated with IVIg for over 10 years (off-label). She was 39 years old when she received the first dose in our clinic. The diagnosis had been confirmed histologically more than 10 years earlier. The patient showed a livedoid, reticular pattern on the legs and forearms, with recurrent, very painful, ulcerations on the lower legs. Therapy attempts were made with low molecular weight heparins, pentoxifylline (600 mg/day for 4 years), etoricoxib (60 mg/day, then every 2–3 days for 3 years), dipyridamole/acetylsalicylic acid (400/50 mg/day for one month, short-term in combination with prazosin), methotrexate and enoxaparin (10 mg/week for one month and 60 mg/day for 4 months), and cyclophosphamide and prednisolone (50 mg/day and 5 mg/day for 5 months). The patient received 1 g/kg IVIg over 2 days initially every 4 weeks, and the symptoms improved quickly for the first time. From the 6th cycle on, she was co-medicated with acetylsalicylic acid (100 mg/day). After the 10th cycle, she received 3 more in a 4–6-month interval and the condition gradually deteriorated again. In addition, polyneuropathy was diagnosed. The intervals between IVIg infusions were shortened again, and acetylsalicylic acid was substituted with clopidogrel (75 mg/day). Under this regime, the inflammation and dysesthesias regressed. In the further course, the intervals of IVIg infusions were slowly extended again up to 4 months. The patient tolerated the infusions well. She reported severe headache only once, lasting for a week and starting 2 days after one of her last IVIg cycles during the observation period.
Another female patient (Fig. 1) with livedoid vasculopathy diagnosed 7 years earlier received 6 doses of 2 g/kg IVIg over 2 days in 4-week intervals at the age of 28 years (off-label), initially in combination with cyclosporine (200 mg/day for one month). Before that, the patient had received repeated doses of intensified prednisolone (1 mg/kg declining dose), as well as cyclophosphamide/dexamethasone (3 cycles of 500/300 mg over 3 days every 4 weeks, followed by 8 cycles of dexamethasone) with prednisolone (10 mg/day) and azathioprine (100 mg/day) as interval therapies, and further methylprednisolone (5 cycles of 750 mg over 3 days every 4 weeks) with as interval therapy cyclosporine (up to 250 mg/day), in addition to anticoagulant therapy. The patient reported headache and nausea as side-effects of IVIg. However, after many years of relapsing-remitting disease course, the skin lesions healed completely with only residual post-inflammatory hyperpigmentation. Following IVIg therapy the patient continued therapy with dalteparin (5,000 IU/day) for another 2.5 years. At the last follow-up without therapy 6 months later, the patient was still in remission.
Fig. 1. A female patient with livedoid vasculopathy (a) 6 months before the first cycle, (b) 3 weeks after the first cycle, and (c) >2 years after 6 treatment cycles with 2 g/kg of body weight intravenous immunoglobulins given over 2 days at 4-week intervals.
IVIg have an immunomodulatory effect with a good tolerability and safety profile. They can be used successfully for the treatment of immune dermatological diseases. Due to the lack of approval, their use is primarily limited to rare refractory diseases and is usually off-label (7). A decision has been taken by the German Federal Joint Committee (G-BA) on the use of IVIg in dermatomyositis. In this case series, we present 32 patients from our clinic with rare immune dermatoses and the course of therapy on IVIg.
The successful and safe treatment of refractory forms of dermatomyositis with IVIg was demonstrated in a randomized controlled study (2). The European guidelines recommend the use of IVIg as a second-line therapy, always in combination with another immunosuppressant (7). While the recommended dose is 2 g/kg administered over 2–5 days, in this case series, we found that a reduced dosage was often as effective as the standard dosage, and better tolerated. There are RCTs also for the treatment of pemphigus vulgaris and bullous pemphigoid with IVIg (5, 6). Several case series confirm the efficacy and tolerability in other blistering autoimmune diseases (8). A comparative study supports the efficacy of IVIg in mucous membrane pemphigoid (9). In alignment with the existing literature, our patients have equally benefited from IVIg therapy. In contrast, in the European guidelines, no clear recommendation can be given for the treatment of systemic sclerosis with IVIg (7). Recent data speak for a safe and successful use (10). In the current study, IVIg had a positive effect on the course of the disease. Also, vasculitis can be successfully treated with IVIg. They are used as first-line therapy for the treatment of Kawasaki syndrome. For all other forms of vasculitis, IVIg are only recommended after failure or contraindication of standard therapy. In fulminant progressive disease courses, early therapy with IVIg is indicated to prevent tissue injury (7, 11). The presented patient, with necrotizing vasculitis and an acute danger to the foot, received IVIg and showed a positive response despite the advanced disease. Nevertheless, despite the fulminant, therapy-resistant course, IVIg were not approved by the health insurance. Treatment of urticarial vasculitis is generally difficult (12). The patient from the current cohort also showed a chronic relapsing disease course. IVIg therapy can be attempted, but the likelyhodd of a response is not clear. The use of IVIg for the treatment of Stevens-Johnson syndrome/toxic epidermal necrolysis is controversial (13, 14). The European guidelines recommend an early application and, unlike other diseases, a unique, high-dose administration of ≥ 3 g/kg over 3–5 days (7). How far IVIg are actually effective in this disease is not clearly determined in the literature. Our patient with Stevens-Johnson syndrome had progressive skin lesions and a pronounced feeling of illness, despite high doses of prednisolone. After additional administration of 2 g/kg IVIg over 4 days, his condition eventually improved. Treatment of pyoderma gangrenosum with IVIg is an option, particularly in severe cases, but there is little evidence available. It is, therefore, more important to collect data from different clinics (15). Both patients from our cohort with progressive pyoderma gangrenosum on repeated adjuvant IVIg therapy have benefited from it, and existing lesions have been partially healed. The positive effect of IVIg in therapy-resistant livedoid vasculopathy is described in the literature (16, 17). The association with peripheral neuropathy is also known (18). In one of our patients with long-term treatment with 1 g/kg IVIg over 2 days up to 4 months, both dermatological and neuropathic symptoms were controlled. In the second patient, 6 cycles of 2 g/kg IVIg led to complete, long-lasting remission.
In conclusion, the patients in our case series have benefited, for the most part, from an adjuvant therapy with IVIg and have shown good tolerability. None of the 32 patients experienced severe side-effects, such as thromboembolic events. Two of the 7 patients who had side-effects did not wish to continue therapy because of headache, abdominal pain and chest pain. The standard dose of 2 g/kg administered over 2–5 days was usually given for 3–6 cycles and, in some cases, reduced to 0.4–1 g/kg and was effective. However, one of the limitations of the current study is the lack of validated clinical scores (such as the Pemphigus Disease Area Index (19)) or simple scoring systems (20) to specify treatment response, due to limited documentation in our medical records. In addition, potential biases include selection bias, because of the single-centre patient population. The decision about if and when to initiate or discontinue IVIg therapy, the number of cycles and different dosages was a result based on discussion between the physician and the patient and the respective health insurance. Moreover, due to the retrospective study design, with no control group and a limited number of patients, these findings may not be generalizable. Nevertheless, since rare diseases are included, we believe that this retrospective case series may be useful for clinicians and patients, as it confirms previous results and suggests that, despite high treatment costs, IVIg therapy may be a valid option for the treatment of rare immune dermatological diseases for which the standard immunosuppressive therapy was not sufficiently effective. At a given indication and disease and treatment course, reimbursement should be requested from the respective health insurance.
Conflicts of interest. RS received a travel grant from ALK-Abelló Arzneimittel GmbH. KM received honoraria or travel expenses for lecture and research activities from Biogen GmbH, AbbVie Deutschland GmbH & Co, Celgene GmbH, Janssen-Cilag GmbH, and UCB Pharma GmbH. KG received consulting fees from Baxalta/Baxter. MW received speakers honoraria for advisory boards and lecture activities from Mylan Germany GmbH, ALK-Abelló Arzneimittel GmbH, Allergopharma GmbH & Co. KG, HAL Allergie GmbH, Stallergenes GmbH, Bencard Allergie GmbH, DBV Technologies SA, Novartis AG, Aimmune Therapeutics UK Ltd, Sanofi-Aventis Deutschland GmbH, Regeneron Pharmaceuticals, Inc., Biotest AG, and Boehringer Ingelheim Pharma GmbH & Co. KG.


 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize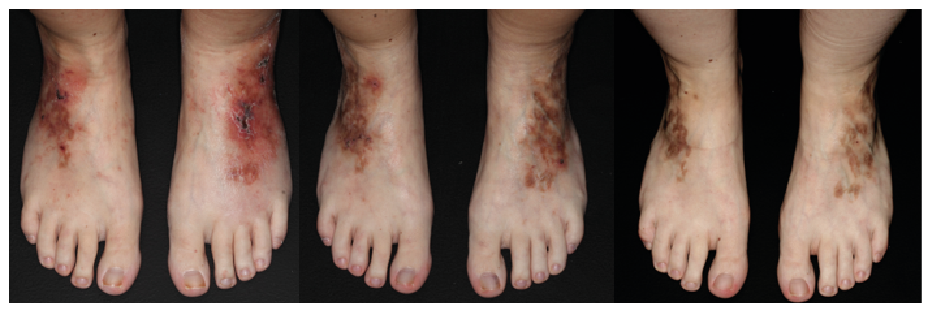 Click to show fullsize
Click to show fullsize