


1Department of Dermatology, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya 466-8550 and 2Department of Genetics, Research Institute of Environmental Medicine, Nagoya University, Nagoya, Japan. *E-mail: takeichi@med.nagoya-u.ac.jp; makiyama@med.nagoya-u.ac.jp
Accepted Feb 2, 2021, Epub ahead of print Feb 8, 2021
Acta Derm Venereol 2021; 101: adv00423.
doi: 10.2340/00015555-3764
Porokeratosis is a genetically heterogeneous disorder that can be caused by mutations in any of the 4 genes involved in the mevalonate pathway (MVK, MVD, PMVK and FDPS) or by SLC17A9, which encodes solute carrier family 17 member 9 (1). Porokeratosis is a skin-specific autoinflammatory disease that is often inherited and is linked to ultraviolet light exposure and immunosuppression (2). Bullous pemphigoid (BP) is characterized clinically by tense blisters on the entire body, and histopathologically by subepidermal blisters with eosinophilic infiltration (3). To date, porokeratosis in association with BP has not been reported, to our knowledge. We report here 2 patients with porokeratosis carrying MVD mutations and in association with BP.
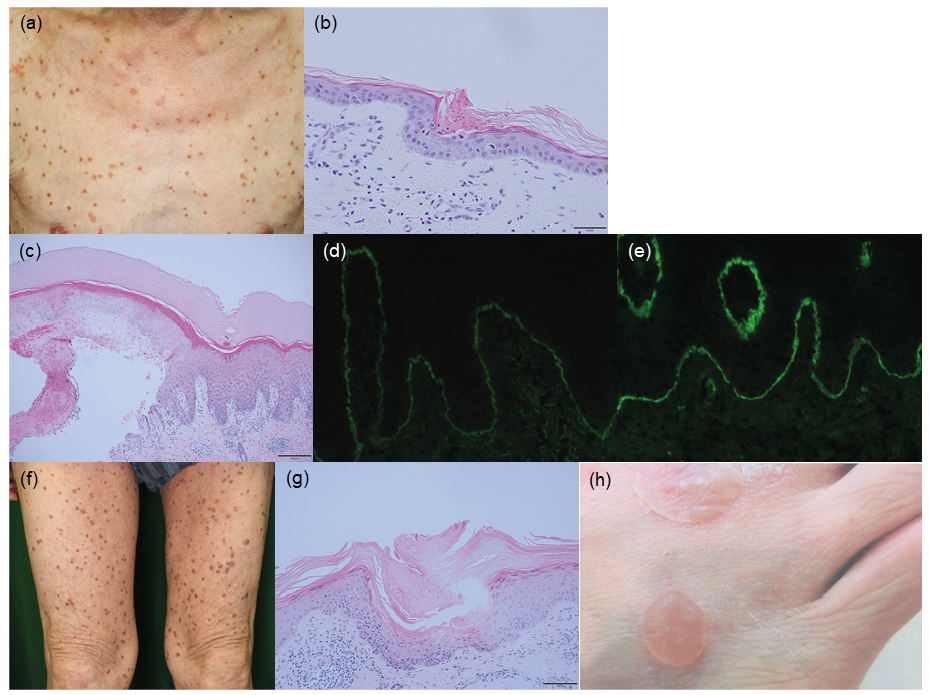
Case 1. A 78-year-old man presented with small, round plaques with well-defined boundaries on his extremities and trunk (Fig. 1a). The eruptions were histopathologically diagnosed as porokeratosis (Fig. 1b). His medical history included atrial fibrillation, hyperuricaemia and hypertension. When the porokeratosis first occurred on his leg, he was treated with apixaban, olmesartan medoxomil and febuxostat. Two years after the onset of porokeratosis, he presented with tender blisters on the back of his left foot for the first time, which spread all over his body. Circulating anti-BP180 antibody was detected by enzyme-linked immunosorbent assay (ELISA) (index: 60.3; cut-off < 9). A skin biopsy specimen from a blister showed a subepidermal cleft with minimal inflammatory infiltrates and perivascular infiltration of lymphocytes and eosinophils in the dermis (Fig. 1c). Direct immunofluorescence staining of the skin biopsy specimen revealed linear deposition of IgG and C3 in the epidermal basement membrane zone (Fig. 1d, e). His BP disease area index (BPDAI) was 9/120.
Fig. 1. Clinicopathological features of the 2 patients. Case 1: (a) Multiple keratotic plaques on the chest. (b) Haematoxylin-eosin staining of a skin biopsy sample from a hyperkeratotic lesion shows hyperkeratosis and cornoid lamella without granular layers. (c) Haematoxylin-eosin staining of a skin biopsy sample from a bulla shows a subepidermal cleft with minimal inflammatory infiltration in the basement membrane zone, and perivascular eosinophilic and lymphocytic infiltration in the superficial dermis. (d, e) Results of direct immunofluorescent staining for (d) IgG and (e) C3 in skin biopsy samples. Scale bars: (b) 50 um, (c) 200 um. Case 2: (f) Keratotic plaques and annular papules on the thighs. (g) Histopathologically, hyperkeratosis, cornoid lamella and a lack of granular layers are seen in the epidermis. Scale bar: 100 μm. (h) Tense bullae on the dorsum of the right foot.
Case 2. A 73-year-old man presented with keratotic plaques on the trunk and extremities (Fig. 1f). A skin biopsy specimen from a keratotic plaque showed cornoid lamella (Fig. 1g). He did not have any past history of illness or medications. One year after first appearance of the porokeratosis, he also developed blisters on the dorsum of both feet (Fig. 1h). A skin biopsy from a blister showed a subepidermal cleft with eosinophilic infiltration. Direct immunofluorescence staining of blistered skin revealed linear deposition of IgG and C3 in the epidermal basement membrane. His BPDAI was 31/120.
This study was performed according to the principles of the Declaration of Helsinki and was approved by the ethics committee of Nagoya University Graduate School of Medicine. Written informed consent was obtained from both patients. Whole-exome sequencing of genomic DNA from peripheral blood samples of the 2 patients revealed previously reported non-synonymous heterozygous variants in MVD, c.1A>G (p.Met1?) in case 1 and c.746T>C (p.Phe249Ser, rs761991070) in case 2 (1, 4), which were confirmed by Sanger sequencing. No potentially pathogenic mutations were identified in any other genes implicated in porokeratosis.
Recently, second-hit somatic mutations were identified in disseminated superficial actinic porokeratosis and linear porokeratosis (5). The mevalonate pathway is essential for cell growth and differentiation, gene expression, cytoskeleton assembly, and post-translational modification of proteins involved in intracellular signalling (6). These facts suggest that individual lesions in various porokeratosis variants arise in regions affected by second-hit mutations in the genes encoding key components of the mevalonate pathway (6, 7).
The mutation c.1A>G (p.Met1?) in case 1 was reported in 3 independent families in 2 reports (1, 4). Although c.1A>G is in a translation initiation codon, there are no reports describing an aberrant translation initiation site. Thus, according to the recommendation of the HGVS nomenclature, the consequence, at the protein level, of a variant cannot be predicted (i.e. it is unknown) (8).
The mutation c.746T>C (p.Phe249Ser) in case 2 seems to be the most common mutation in MVD and is possibly a hot-spot mutation (1, 4). According to the 3-dimensional structures of the missense variations predicted using I-TASSER, the p.Phe249Ser mutation results in the loss of a hydrogen bond with phenylalanine252, and the hydrogen bonds with alanine253 and arginine247 are lengthened and shortened, respectively, compared with those of the wild type (1). However, it has not been revealed whether this variation produces a functional change in MVD in vivo (1). The age of onset in patients with mutations in MVD shows a wide range (1). In addition, incomplete penetrance of porokeratosis associated with the MVD mutation c.746T>C (p.Phe249Ser), which was detected in case 2, was reported previously (1). These findings suggest that a specific trigger, inducing second-hit mutations in MVD, may be necessary for the onset of porokeratosis.
Immunosuppression-induced porokeratosis has been reported previously, and immunosuppression might be a possible inducer of second-hit mutations in porokeratosis-causative genes (9, 10). However, the 2 patients reported here did not take any immunosuppressants until the occurrence of porokeratosis. In the current 2 cases, widespread porokeratosis was present 1–2 years before the onset of BP blisters. In addition, in neither patient did BP blisters appear on skin regions where porokeratosis plaques had been observed. This study cannot exclude the possibility that the occurrence of porokeratosis with BP in the current patients was merely coincidental. Further studies of similar cases are needed to confirm the relationship between porokeratosis and BP.
This research was supported by the Japan Agency for Medical Research and Development (grant number JP19ek0109281h0003) to MA. The study was supported in part by Japan Society for the Promotion of Science KAKENHI (grants number 18H02832) to MA and 20K08648 to TT. The study was also supported in part by The Nakatomi Foundation, grants from the Maruho Takagi Dermatology Foundation, and JSID’s fellowship Shiseido Research Grant 2019 to TT.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize