


Department of Dermatology, University of Heidelberg, DE-69120 Heidelberg, Germany. E-mail: kai-philipp.linse@med.uni-heidelberg.de
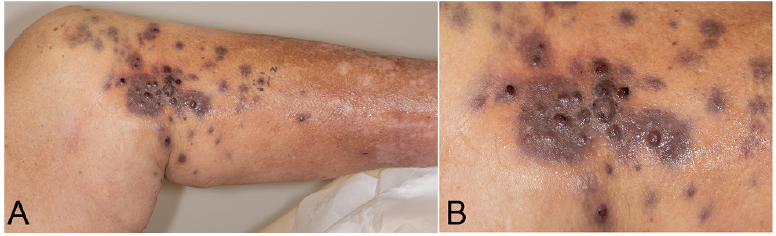
A 71-year-old male patient presented at the dermatology department with multiple firm livid cutaneous nodules on the proximal lower leg, scattered over an area approximately 15 × 25 cm (Fig. 1). The lesions initially appeared six weeks before presentation; they were painful on palpation and progressive in number and size. In addition, clinical examination and ultrasound scan revealed a firm subcutaneous lesion, approximately 5 cm diameter, in the affected area, and a superficial venous thrombosis of the adjacent great saphenous vein. Two punch biopsies of the cutaneous nodules, for histopathological evaluation, and a magnetic resonance imaging (MRI) scan were performed.
Histopathology (Fig. 2A, B) showed a dermal, highly pleomorphic, heterochromatic tumour with high cell density, intense vascularization and extravasation of erythrocytes. The tumour cells had a varying size of nuclei and a high number of atypical mitotic figures. Immunohistochemistry (Fig. 2C–E) showed nuclear expression of ETS transcription factor ERG (ERG), cytokeratin expression (AE1/AE3), positivity for cluster of differentiation 31 (CD31) and overexpression of MYC proto-oncogene (MYC). Furthermore, there was high proliferative activity (Ki67: 70%) and the tumour stained negative for calmodulin binding transcription activator 1 (CAMTA1), prostate specific antigen (PSA), S100 calcium binding protein (S100) and podoplanin (D2-40).
The MRI scan showed a subcutaneous lesion, approximately 4.9 × 3.6 cm, medial to the popliteal area, and two more subcutaneous lesions, 14 mm and 7 mm diameter, in the upper thigh.
What is your diagnosis? See next page for answer.
Fig. 1. Multiple livid nodules on the left proximal lower leg. (A) Overview, (B) close-up view.
Fig. 2. Diffusely infiltrating dermal tumor with high cell density, intense vascularization and extravasation of erythrocytes. (A: Haematoxylin eosin (HE) x50). The tumor cells exhibit large, atypical, pleomorphic nuclei, vacuolated cytoplasm and a high number of atypical mitotic figures. (B: HE x200). Immunohistochemistry revealed intense positivity for CD31 (C: CD31 x50) and a high proliferative activity (D: Ki67 x50). CAMTA1 staining was negative (E: CAMTA1 x100).
Acta Derm Venereol 2021; 101: adv00490.
Diagnosis: Epithelioid angiosarcoma with cutaneous metastases
Clinico-pathological correlation was consistent with a diagnosis of an epithelioid angiosarcoma with cutaneous metastases. Epithelioid angiosarcoma (EA) is a rare and highly malignant neoplasm of endothelial cells with predominantly epithelioid appearance. EA is most commonly located in the soft tissue of the extremities, but it can also be found in the skin, thyroid gland, adrenal glands and bone. EA has a predominance for males and is seen mainly in older patients, with a peak incidence in the seventh decade of life (1–3).
EA has a wide range of clinical presentations, depending on the location. The most prevalent symptoms are an enlarging and painful mass, as well as recent haemorrhage or a coagulopathy. As a high-grade sarcoma, EA is associated with a poor prognosis and early metastases, most frequently to the lungs, lymph nodes, soft tissue, bone, and liver (2, 3).
Histopathology shows sheets and nests of large, pleomorphic, round to polygonal epithelioid cells. The cells are rich in eosinophilic cytoplasm, have vesicular nuclei and prominent nucleoli. Areas of necrosis, haemorrhage and numerous mitotic figures are common. Intracytoplasmic vacuoles containing erythrocytes and a rudimentary vasoformative architecture are frequently, but not always, present. Immunohistochemistry in EA reveals positivity for vimentin, shows high levels of Ki67, and is characteristically positive for endothelial markers, including CD31, ERG, coagulation factor VIII (FVIII), and friend leukemia integration 1 transcription factor (FLI1) (2).
EA can be mistaken for epithelioid haemangioendothelioma (EHE). The neoplastic cells of EA are usually larger and more atypical than those of EHE and show a higher mitotic activity. Furthermore, single cell necrosis or large areas of mass necrosis are more frequently observed in EA than in EHE. Cytogenetic studies revealed that the t(1;3) (p36;q23-25) translocation, creating the WW domain containing transcription regulator 1 (WWTR1)-calmodulin-binding transcription activator 1 (CAMTA1) fusion gene, is unique to EHE and can be found in approximately 85% of cases of EHE.
Antibodies directed against CAMTA1 may be used for immunohistochemistry to detect the WWTR1-CAMTA1 fusion gene with 85% sensitivity and nearly 100% specificity within the differential diagnosis of epithelioid soft tissue tumours, including EA (3).
Further differential diagnoses include metastatic carcinomas, malignant mesothelioma, melanoma, anaplastic lymphoma, epithelioid peripheral nerve sheath malignancies, and epithelioid sarcoma, which can be differentiated with a panel of immunohistochemical stains (4).
Treatment options for limited or locally advanced disease consist of surgical extirpation of the primary tumour followed by radiotherapy (2). Advanced stages may profit from taxane- or doxorubicin-based chemotherapeutic regimens (5, 6). Nevertheless, prognosis in general is very limited, and patients of advanced age, with larger tumour size, retroperitoneal tumour location or Ki67 values over 10%, have a considerably poorer prognosis (4).
Tumour mutation analyses revealed a whole gene deletion of the cyclin dependent kinase inhibitor 2A (CDKN2A) gene, and chemotherapy with paclitaxel (80 mg/m2) weekly was started.
We thank Professor Thomas Mentzel (Dermatopathology Friedrichshafen, Germany) for reviewing the slides and providing the CAMTA1 staining.


 Click to show fullsize
Click to show fullsize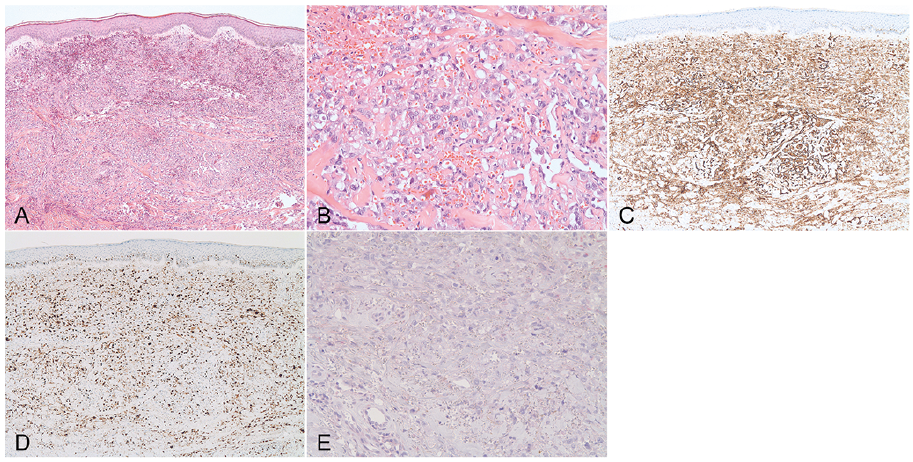 Click to show fullsize
Click to show fullsize