


1Skin Research, LEO Pharma A/S, Ballerup, Denmark, 2Department of Dermatology, Venereology, and Allergology, University Hospital Schleswig-Holstein, Campus Kiel, Kiel, 3Division of Immunodermatology and Allergy Research, Department of Dermatology, Allergology, and Venereology, Hannover Medical School, Hannover, Germany and 4Center for Evidence-based Health Care (ZEGV), Medical Faculty Carl Gustav Carus, TU Dresden, Dresden, Germany. *E-mails: hnddk@leo-pharma.com; sweidinger@dermatology.uni-kiel.de
Accepted Apr 22, 2021; Epub ahead of print Apr 26, 2021
Acta Derm Venereol 2021; 101: adv00447.
doi: 10.2340/00015555-3810
Atopic dermatitis (AD) is a chronic, pruritic skin disease characterized by type 2 immune-mediated inflammation and skin barrier dysfunction (1). The impaired skin barrier in AD is related to decreased expression of epidermal barrier proteins and to changes in lipid composition in the stratum corneum. AD lesions are characterized by increased expression of pro-inflammatory mediators, such as chemokines, leading to recruitment of immune cells, which may further exacerbate the inflammation.
Interleukin (IL)-13 and IL-4 are the signature type 2 cytokines involved in AD. These cytokines both signal through the type II receptor, composed of IL-4Rα and IL-13Rα1, and display functional redundancy in cells harbouring this receptor, including keratinocytes and fibroblasts. This is in contrast to cells, such as T lymphocytes, containing the type I receptor (IL-4Rα combined with the common gamma chain), which is only used by IL-4 for signalling. Monoclonal antibodies targeting IL-4Rα (dupilumab) or IL-13 (tralokinumab and lebrikizumab) have demonstrated clinical efficacy in AD (2–4). Interestingly, protein levels of IL-13, but not IL-4, are consistently detected and shown to be increased in AD skin across studies (5, 6). Furthermore, 3 recently published studies using RNA-sequencing of AD biopsies found increased levels of IL13 in lesional and non-lesional AD skin, whereas expression of IL4 was undetectable or very low (7–9). Independent studies also found a correlation between expression levels of IL-13 at mRNA or protein level in lesional AD skin and disease severity (6, 7).
The aim of the current study is to further investigate effects of IL-13 on AD-associated genes in human skin cells and to provide molecular insights into the mechanism of action of tralokinumab, a fully human IgG4 monoclonal antibody that specifically neutralizes IL-13.
Detailed descriptions of cell cultures, cytokine stimulation, gene expression analysis, protein analysis and data analysis are shown in Appendix S1.
Correlation of IL13 expression with genes expressed in skin biopsies from patients with moderate-severe AD was evaluated by combining data from 3 recent transcriptomic studies (7–9). The combined datasets include RNA-seq data from chronic lesions from each patient (n = 89) with paired non-lesional samples (n = 87), and, in addition, acute lesions from a small subgroup of the patients (n = 11). A positive correlation was found in lesional AD skin between expression of IL13 and several pro-inflammatory mediators as well as IL13RA2 (Table I and Fig. S1). In contrast, a negative correlation was seen in lesional AD skin between expression of IL13 and genes related to skin barrier function. For most of the genes, the correlation with IL13 was even more pronounced when paired lesional and non-lesional samples were included in the analysis (Table I).
Table I. Expression of IL13 correlates with several inflammation and epidermal barrier associated genes in skin biopsies from patients with moderate-severe atopic dermatitis
Using cultures of human skin cells, IL-13-mediated regulation of several of these genes was then explored, as well as their modulation by tralokinumab. Primary human epidermal keratinocytes (HEK) and human dermal fibroblasts (HDF) were stimulated with 10 ng/ml (0.8 nM) and 2 ng/ml (0.16 nM) IL-13, respectively, each corresponding to ~80% of maximal gene responses, in the presence of a broad range of concentrations (0.003–30 nM) of tralokinumab or isotype control antibody for 24 h. Using qPCR analysis, the effects of IL-13 and tralokinumab were evaluated on gene expression of CCL2, CCL17, CCL22, CCL26, NTRK1 and IL13RA2 in HEKs, and of CCL2, CCL11, CCL17, CCL22 and POSTN in HDFs.
In line with the correlation analysis, IL-13 markedly induced gene expression of CCL2, CCL26, NTRK1 and IL13RA2 in keratinocytes (Fig. 1a) and CCL2, CCL11 and POSTN in dermal fibroblasts (Fig. 1c). Tralokinumab, but not the isotype control antibody, potently neutralized IL-13, resulting in a dose-dependent and full inhibition of the inflammatory markers, with sub-nanomolar IC50 values (Fig. 1a, c and Table SI). As IL13RA2 is directly induced by IL-13 in keratinocytes, levels of IL13RA2 are expected to be decreased in AD skin upon inhibition of the IL-13 signalling axis, as is also shown by the IL-4Rα-blocking antibody dupilumab (10). In addition to gene expression analysis, secreted levels of CCL-2/MCP-1 were determined by enzyme-linked immunoassay (ELISA). Here, it was confirmed that IL-13 induced secretion of CCL-2/MCP-1 from HEKs and HDFs, which was potently and dose-dependently inhibited by tralokinumab, with IC50 values of 217 and 336 pM, respectively (Fig. 1b, d and Table SI). IL-13 did not induce expression of CCL17 and CCL22 in HEKs and HDFs. However, IL-13 induced secretion of CCL-17/TARC and CCL-22/MDC from human peripheral blood mononuclear cells (PBMCs), which was potently and dose-dependently inhibited by tralokinumab (Fig. S2 and Table SI).
Fig. 1. Atopic dermatitis (AD)-associated genes are regulated by interleukin (IL)-13 and modulated by tralokinumab in cultures of human skin cells. Cells were stimulated with IL-13 in absence or presence of a broad range of tralokinumab or IgG4 isotype control antibody for 24 h and subjected to gene expression analysis by qPCR (a, c and e) and protein analysis of CCL-2 in supernatants by enzyme-linked immunoassay (ELISA) (b and d). (a) Gene expression analysis on primary human epidermal keratinocytes (HEK). (b) CCL-2 levels (pg/ml) in supernatants from HEK cells. Dashed line indicates the lower limit of detection. (c) Gene expression analysis on primary human dermal fibroblasts (HDF). (d) CCL-2 levels (pg/ml) in supernatants from HDF cells. (e) As in (a), however, using CaCl2 differentiated HEK cells. Gene expression data is shown as fold change compared with the untreated controls (green symbol).
In addition, the effects of IL-13 and tralokinumab were investigated on 5 skin barrier related genes, FLG, FLG2, LOR, ELOVL3 and ELOVL6, that had shown a negative correlation with IL13 (Table I and Fig. S1). Differentiated primary human epidermal keratinocytes were stimulated with 50 ng/ml (4.2 nM) IL-13 in the presence of a broad concentration range (0.78–200 nM) of tralokinumab or isotype control antibody for 24 h. In alignment with the correlation analysis and with previous studies (reviewed in (6)), IL-13 clearly downregulated expression of the skin barrier related genes (Fig. 1e), with the exception of ELOVL6, of which expression levels remained unchanged upon stimulation with IL-13. Treatment with tralokinumab potently neutralized IL-13, leading to a dose-dependent and full normalization of FLG, FLG2, LOR, and ELOVL3 expression, with low nanomolar IC50 values (Table SI).
In summary, skin barrier- and inflammation-associated genes in AD skin correlate with IL13 expression, and a subset of these genes were investigated and shown to be regulated by IL-13 and fully normalized by tralokinumab in vitro. Compared with estimated steady-state concentrations for tralokinumab of approximately 30–35 nM in skin of patients with AD after 300 mg once every 2 weeks (Q2W) dosing, the IC50 values from the in vitro studies are at clinically relevant concentrations and the potency of tralokinumab may probably be underestimated due to the high concentrations of IL-13 used in vitro. These data provide molecular insights and may explain the clinical effects of tralokinumab, including skin barrier restoration and reduction in skin inflammation.
In vitro studies and re-analysis of RNA-sequencing data were funded by LEO Pharma.
Conflicts of interest. MT, TL and HN are employees of LEO Pharma. SW is a speaker, advisory board member and/or investigator for AbbVie, Galderma, Incyte, Kymab, La Roche-Posay, LEO Pharma, Lily, Novartis, Pfizer, Regeneron and Sanofi-Genzyme. TW is a speaker, advisory board member and/or investigator for Abbvie, Celgene Janssen, Galderma, LEO Pharma, Sanofi-Genzyme, and Novartis. JS received institutional funding for IITs by Sanofi, Pfizer, ALK, Novartis, and served as an advisor for Novartis, Sanofi and Lily. The other authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize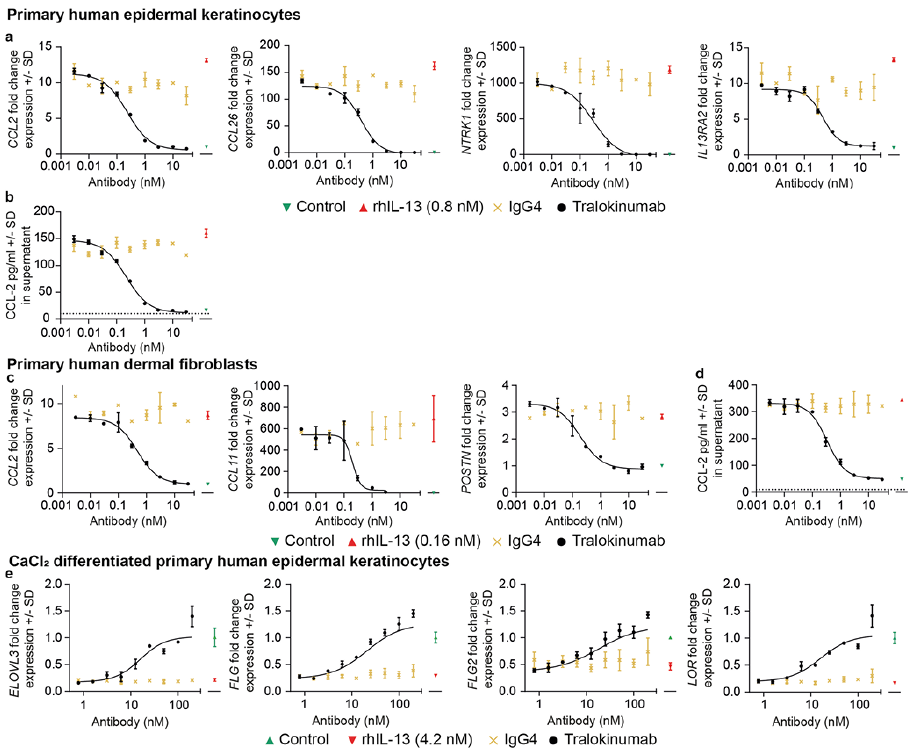 Click to show fullsize
Click to show fullsize