


Department of Dermatology, Institute of Medical Sciences, Medical College of Rzeszow University, ul. Fryderyka Szopena 2, PL-35-055 Rzeszów, Poland. *E-mail: adi_medicalis@go2.pl
A 26-year-old man presented to our department with a 3-week history of pruritic annular, urticarial-like plaques scattered over his neck, trunk and extremities. The lesions were refractory to previous treatment with 0.7 mg/kg/day oral prednisone, clemastine and aciclovir. The patient did not report systemic symptoms, neither had he taken any other medications over the preceding weeks. His medical and family history was unremarkable.
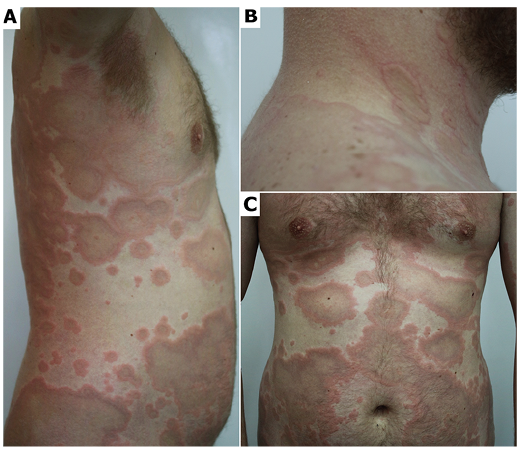
Clinical examination revealed well-demarcated annular erythematous plaques with central hyperpigmentation and slightly raised borders located predominantly on the trunk and proximal extremities (Fig. 1). Blood tests revealed leukocytosis (18,280 cells/μl, normal range 4,000– 10,000 cells/μl) with moderate eosinophilia (3,410 cells/μl, normal range < 450 cells/μl). Serum antinuclear antibodies and anti-neutrophil cytoplasmic autoantibodies were negative; complement C3 and C4 levels were normal. Bacterial (Borrelia burgdorferi, Helicobacter pylori, Mycoplasma pneumoniae, Chlamydia pneumoniae), viral (hepatitis B and C, HIV) and parasitic (intestine parasites, Toxocara canis, Trichinella spiralis) infections were excluded.
Imaging tests showed no abnormalities. Identification of FIP1L1-PDGFRA, ETV6-PDGFRB and BCR-ABL fusion genes, along with bone marrow biopsy, were performed, and ruled out the haematological causes of peripheral eosinophilia. Histopathology revealed basal vacuolar changes and perivascular inflammatory infiltrate abundant in eosinophils, with sparse lymphocytes, and histiocytes predominantly in the superficial dermis, in the absence of vasculitis (Fig. 2).
What is your diagnosis? See next page for answer.
Fig. 1. Initial clinical presentation of the patient. (A, C) Multiple ring-shaped polycyclic plaques that heal centrally with hyperpigmentation on the trunk and upper arms. (B) Coalescent annular plaques with markedly elevated borders on the neck and upper back.
Fig. 2. Histopathology of a lesional skin biopsy specimen. (A) A superficial dermal predominantly perivascular inflammatory infiltrate composed of lymphocytes, histiocytes, and abundant eosinophils (×200, haematoxylin and eosin; H&E). (B) A prominent perivascular component of the infiltrate in the absence of vasculitis (×400, H&E).
Acta Derm Venereol 2021; 101: adv00450.
Diagnosis: Eosinophilic annular erythema
On initial examination, several conditions were taken into account in the differential diagnosis, including Wells syndrome, erythema annulare centrifugum, granuloma annulare, subacute cutaneous lupus erythematosus, urticarial vasculitis, multifocal erythema chronicum migrans, and non-bullous phase of bullous pemphigoid. The diagnosis was facilitated by the results of laboratory findings (exclusion of infectious causes, negative immunological profile) and the histopathological features compatible with eosinophilic annular erythema (EAE) (predominantly perivascular infiltrate with abundant eosinophils in the dermis and absence of so-called “flame figures” highly suggestive of Wells syndrome). Once the diagnosis was made, the patient was started on methylprednisolone (24 mg/day) combined with dapsone (100 mg/day) with no improvement after 4 weeks. Subsequent treatment with cyclosporine A (4.5 mg/kg/day) for 3 weeks was also ineffective. Consequently, the patient was started on hydroxychloroquine (200 mg twice daily), which led to a dramatic improvement in the clinical symptoms within 3 weeks.
EAE is a rare, benign dermatosis characterized by recurrent outbreaks of annular urticaria-like lesions and/or erythematous plaques (1). The aetiology of EAE is unknown, although it has been suggested that it develops as a result of a hypersensitivity reaction to an unidentified allergen (2). Some authors consider EAE to be a peculiar variant in the spectrum of Wells syndrome (also referred to as eosinophilic cellulitis), considering that blood eosinophilia and “flame figures” can also be observed in well-developed or long-standing lesions (1, 3). As the diagnostic criteria for EAE have not yet been established, it is presumably underdiagnosed (4). EAE was first described by Peterson & Jarratt in 1981 and involved only children (5). The authors reported infants with erythematous papules and interrupted arcs that rapidly evolved into annular erythema and ring-shaped oedematous plaques, lesions typical of the disease. Twenty years later, the first description of EAE in an adult was published regarding a case of a 62-year-old man with a similar clinical presentation and histological findings to those described previously only in paediatric patients (6).
Typically, patients with EAE present with asymptomatic or mildly pruritic annular urticarial-like lesions and/or erythematous and oedematous ring-shaped plaques, which may coalesce to produce concentric, serpiginous and polycyclic shapes. In most cases trunk and limbs are affected, although, in some cases, lesions may also involve the face. EAE is known for its chronic relapsing and remitting course, and the lesions may persist for months or even years (3).
Histologically, EAE is characterized by: (i) predominantly perivascular lymphohistiocytic infiltrate with a prominent eosinophilic component, (ii) absence of “flame figures”, and (iii) unaltered epidermis. Wells syndrome histology is distinct from that of EAE, and includes an interstitial infiltrate of eosinophils, with lymphocytes and histiocytes, which is usually more prominent in the deep dermis. In addition, eosinophilic-staining “flame figures” consisting of collagen fibres coated with eosinophil granule proteins, although not pathognomonic, are a hallmark of this entity (7). As underlined by El-Khalawany et al. (3), patients with EAE with long-standing disease may exhibit histological similarities to Wells syndrome. Given that the clinical presentation of these 2 uncommon eosinophilic entities is often very similar, in some cases thorough clinicopathological study is necessary to make this subtle distinction.
Even though in some cases EAE is self-limiting, it usually has a chronic course (2). There is no standard treatment for EAE; anti-malarials and systemic corticosteroids are most successfully used in the management, and are therefore considered as first options (8). However, the treatment response is variable and early relapses after discontinuation of therapy are not uncommon. Additional treatment modalities reported to have some efficacy include dapsone, cyclosporine A, indomethacin, nicotinamide, ultraviolet B phototherapy, dupilumab, and mepolizumab, among others (9).
Multiple chronic conditions and infections are reported to have a possible association with EAE, including autoimmune thyroid disease, Lyme disease, Helicobacter pylori infection, diabetes mellitus, hepatitis C infection, chronic kidney disease, autoimmune pancreatitis, and internal malignancies (thymoma, clear cell renal carcinoma, metastatic prostate adenocarcinoma) (10, 11).
In conclusion, this case report emphasizes the importance of including EAE in the differential diagnosis when evaluating patients with unexplained, long-standing or treatment-refractory annular lesions. It also highlights the necessity of a thorough laboratory work-up taking into consideration numerous possible associations of this distinct clinical picture.


 Click to show fullsize
Click to show fullsize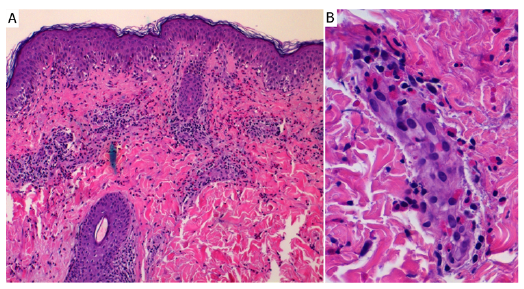 Click to show fullsize
Click to show fullsize