


1Department of Dermatology and Allergy, University Hospital, LMU Munich, Frauenlobstr. 9-11, DE-80337 Munich, Germany and 2Dr Phillip Frost Department of Dermatology and Cutaneous Surgery, Miller School of Medicine, University of Miami, Miami, FL, USA. E-mail: annecharlotte.niesert@med.uni-muenchen.de
Accepted May 26, 2021; Epub ahead of print May 27, 2021
Acta Derm Venereol 2021; 101: adv00472.
doi: 10.2340/00015555-3840
Over the past 6 years, programmed cell death-1 (PD-1) and programmed cell death 1 ligand 1 (PD-L1) antibodies have become the first-line treatments for advanced skin tumours (1). PD-1 and PD-L1 antibodies gradually garnered first- and later-line Food and Drug Administration (FDA) approvals for multiple indications, such as non-small cell lung cancer (NSCLC), renal cell carcinoma, urothelial carcinoma, colorectal and hepatocellular carcinoma, Hodgkin’s lymphoma and, most recently, oesophageal cancer (2). Due to their overall good tolerance and appealing clinical outcome, nivolumab and pembrolizumab have also been approved for adjuvant therapy of malignant melanoma since July 2018 (3). In addition, various clinical trials are investigating the adjuvant use of PD-1 and PD-L1 antibodies in other tumour entities (4–6). Due to the increasing prescription of these drugs, various, previously unknown, immune-related adverse events (irAE) are gaining importance in everyday clinical practice, including cutaneous side-effects in up to 40% of treated patients (7). Thus, patients receiving the treatment in an adjuvant setting deserve profound information and require a thorough risk-benefit assessment before initiation of the therapy.
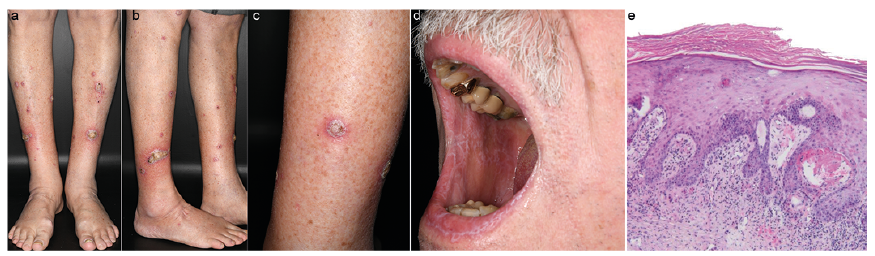
We report here the case of a 77-year-old man who underwent treatment for stage IIIC (American Joint Committee on Cancer 2016) malignant melanoma. He received adjuvant immunotherapy with pembrolizumab, at a dose of 200 mg/kg body weight every 3 weeks, as recommended by an interdisciplinary tumour board. After 6 months of immunotherapy, the patient presented with hyperkeratotic nodules and plaques, up to 10 cm in diameter, on both lower legs (Fig. 1a–d). In addition, typical Wickham striae were found bilaterally on the buccal mucosa. A diagnosis of hypertrophic lichen planus (LP) was confirmed histologically (Fig. 1e). A therapy including topical steroids and keratolytic ointment was initiated. With the patient’s informed consent, continuation of adjuvant therapy with pembrolizumab was decided. Regular staging examinations showed stable tumour presence. However, skin changes progressed towards extensive ulcerated LP (Fig. S1). Therefore, systemic steroids, 1 mg/kg body weight, were added to the topical therapy, and thereafter gradually reduced over a period of 4 weeks. In the following 6 months, close staging examinations continued to show no evidence of pathology. Likewise, skin changes were controlled with low-dose systemic steroid therapy, 10 mg/day.
Fig. 1. (a–c) Both lower legs are affected, with sharply defined, yellowish, hyperkeratotic noduli and plaques with a diameter of up to 4 cm and an erythematous border. (d) Wickham striae at the buccal mucosa. (e) Orthohyperkeratosis over hypergranulosis. Hyperplasia of rete ridges with intensive interface changes. Band-like lymphatic infiltrate with basal vacuolar degeneration of single keratinocytes. Hematoxylin-eosin stain, original magnification x 100.
The electronic databases MEDLINE via Ovid (from 1968), Embase via Ovid (from 1974) and PubMed were searched with the terms “lichen planus”, “checkpoint inhibitor therapy” and “immunotherapy” to identify all relevant records up to 4 September 2020.
Two authors (ACN and OS) independently screened the titles and abstracts for eligibility, which were identified in the database search. For records that were considered relevant according to title and abstract screening, full-text articles were obtained and checked for inclusion and exclusion criteria. The inclusion criteria included case reports of lichen planus that developed during immunotherapy with a PD-1 inhibitor. The PD-1 inhibitor had to have been given adjuvantly or therapeutically for malignancy. The diagnosis had to be confirmed by histology. Whenever discrepancies arose, resolution was achieved by discussion with a third independent author (AG).
The literature search identified 129 references. After removing duplicates, 77 records remained. Following title and abstract screening, 52 references were excluded as they did not meet the inclusion criteria. Full text was not accessible in 2 cases. The 23 remaining records underwent full-text review and 4 were excluded because LP was not histologically proven. The current patient was included as the 20th case in the analysis of immune-related LP.
Analysis of the literature for immune checkpoint-inhibitor-related LP (irLP), revealed a balanced sex distribution, with 50% females, compared with 61% for idiopathic LP. Mean age of onset for irLP was 66.6 years, compared with 46.4 years for idiopathic LP (Table I) (1, 8). The difference can be explained by the higher mean age of tumour patients compared with the general population affected by LP.
Table I. Characteristics of the reviewed cases with immune- related lichen planus (LP)
In idiopathic LP classic cutaneous (88%) and hypertrophic subtypes (7%) are most common, hypertrophic and bullous types of LP occurred more frequently in irLP (Table I) (8). This discrepancy might be due to the lower threshold for reporting pronounced skin findings, such as hypertrophic or bullous LP, compared with discrete skin findings, such as a classic cutaneous LP, or LP as an immune checkpoint-inhibitor-related phenomenon. Previous studies indicate that irLP is reported infrequently and might therefore be underdiagnosed (3, 7, 9).
Most cases were cured with topical or systemic steroids, only a few needed an escalation of therapy with addition of systemic retinoids, dapsone or rituximab (Table I). These results are in line with recent recommendations for severity-adapted therapy of immune-related cutaneous adverse events (irCAE) (10). The proportion of patients with irLP (60%) requiring systemic therapy was higher than that of patients with idiopathic LP (32%) (8). However, as described above, this difference may also be due to the bias of reporting preferential severe cases.
Chronologically, LP appeared a mean of approximately 17 weeks after starting immunotherapy compared with other irCAE, which appear much earlier, after a mean of 3.6 weeks of therapy (7, 9, 11). The onset of irLP is therefore more in line with lichenoid skin reactions to immune checkpoint-inhibitor therapy, which usually appear 6–12 weeks after the onset of therapy (7, 9). Overall, irAE have a rather delayed appearance compared with drug side-effects in general (12).
The difference in distribution between anti-PD-1 and anti-PD-L1 therapy, underlying types of tumour and indications (metastatic vs adjuvant therapy) is due to the widespread use as well as the earlier FDA/EMA approval of, and broader indications for, anti-PD-1 therapy compared with anti-PD-L1 therapy (Tables I and SI) (2).
In 11/20 (55%) cases, anti-PD-1 or anti-PD-L1 therapy was continued despite the irCAE LP. In 9/20 (45%) patients, immunotherapy was terminated due to tumour progression or other irAE. As stated by di Lorenzi et al. (13), this observation indicates that cessation of anti-PD1 and anti-PD-L1 therapy is not mandatory when LP occurs, since intensive topical corticosteroid therapy and systemic corticosteroid treatment offer an effective approach to the control of irLP.
In 12/15 (80%) cases, stable disease or a partial/complete response, and, in 3/15 (20%) cases, progression of the tumour disease was reported (Table I). This data is supported by the meta-analysis of Cortellini et al. (14) demonstrating a statistically significant overall survival benefit and longer progression-free survival in patients presenting with irAE.
This review is limited by the relatively small number of cases. Long-term data regarding the course of both LP and tumour disease are lacking. Further studies with pooled data of larger cohorts of patients with immunotherapy are required to better define the occurrence of LP with respect to tumour disease, duration of PD-1/PD-L1-therapy and tumour outcome.
These data provide evidence that immunotherapy does not have to be discontinued if LP occurs, as the irCAE responded well to topical and systemic corticosteroid therapy. However, detailed information prior to treatment initiation concerning relevant risks and side-effects of immune-checkpoint inhibitor therapy is mandatory for informed consent, especially in the case of an adjuvant treatment approach, as irCAE may have a significant impact on the patient’s quality of life, as demonstrated in the current case.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize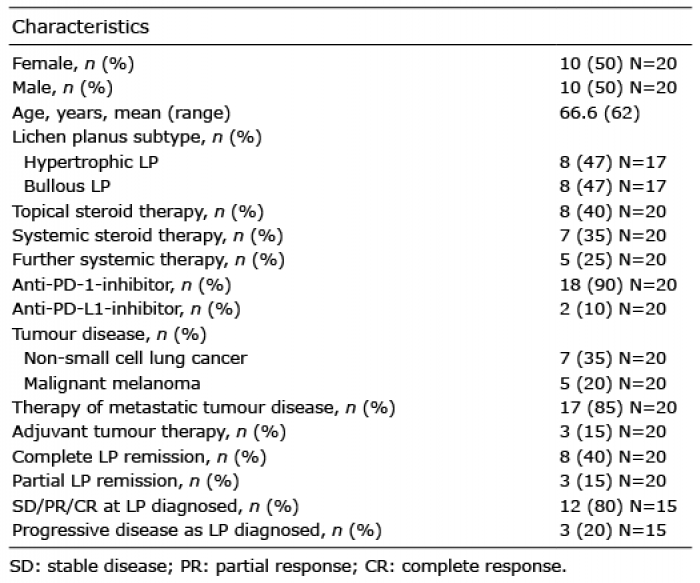 Click to show fullsize
Click to show fullsize