


1Faculty of Health Sciences, University of Southern Denmark, Odense, 2Department of Plastic and Breast Surgery, Aarhus University Hospital, Aarhus, 3Department of Clinical Genetics and 4Department of Dermatology and Allergy Centre, Odense University Hospital, Odense, Denmark
Genodermatoses are inherited syndromes with cutaneous manifestations. Some genodermatoses are associated with malignancy of internal organs and tissues. Early detection of the typical signs of these syndromes is important, because those lesions are a sign of underlying predisposition to extracutaneous neoplasms. The dermatologist has an important role in the early detection of these signs and syndromes, as early detection may affect the clinical course of the disease. We report here the characteristic cutaneous findings that dermatologists should be aware of in order to identify a genodermatosis with a possible associated malignancy. An updated overview of the pathogenesis and clinical findings of these syndromes is provided. Furthermore, surveillance protocols and treatment recommendations are explored.
Key words: genodermatoses; familial cancer; hereditary neoplastic syndromes; genetic predisposition; skin diseases; cutaneous lesions.
Accepted June 21, 2021; Epub ahead of print Jun 23, 2021
Acta Derm Venereol 2021; 101: adv00505.
doi: 10.2340/00015555-3852
Corr: Anne-johanne Andersen, Department of Emergency Medicine, Odense University Hospital, Odense, Denmark. E-mail: Anne-Johanne.Andersen2@rsyd.dk
This article reviews the inherited skin syndromes associated with malignancy of internal organs and tissues. The dermatologist has an important role in the early identification of these syndromes, which may affect the clinical course of the disease. We also provide an up-to-date overview of the pathogenesis, clinical findings, surveillance protocols and treatment of genodermatoses associated with internal malignancy.
Genodermatoses are inherited syndromes with cutaneous manifestations. Some genodermatoses are associated with malignancy of internal organs and tissues. The genetic basis and the clinical phenotype has been further characterized in recent years. Early recognition of the cutaneous signs of these disorders provides affected patients with surveillance and screening regimens, which may positively affect their outcome.
The aim of this review is to outline the characteristic cutaneous findings that dermatologists should be aware of in order to identify a genodermatosis with a possible associated malignancy. An up-to-date overview of the pathogenesis, clinical findings, diagnostic criteria, treatment and management of these genodermatoses is provided.
The earliest historical evidence of neurofibromatosis type 1 (NF1), formerly known as von Recklinghausen’s disease, first appeared in the 13th century. However, it took until 1882, when Friedrich Daniel von Recklinghausen published his landmark paper on the multiple fibromas of the skin and their relationship to the multiple neuromas, for NF1 to be recognized as a separate disorder (1). Research into NF1 was stimulated by the erroneous diagnosis of Joseph Merrick “the elephant man”, whom it was assumed had NF1. It has since been clarified that he is more likely to have had Proteus syndrome. Patients with Proteus syndrome are mosaics, with a somatic disease-causing variant in AKT1 (2).
Molecular genetics and pathophysiology
NF1 is an autosomal dominant disorder, with nearly 50% of cases arising from sporadic mutations. NF1 affects approximately 1 in 2,500–3,000 people (3). By the age of 20 years, penetrance is almost 100%, although expressivity is highly variable (4). NF1 is caused by disease-causing variants in the NF1 gene, a tumour-suppressor gene located on chromosome 17q11.2, encoding neurofibromin. Neurofibromin is a negative regulator of the Ras proto-oncogene, which serves as a signalling molecule for cell proliferation and differentiation (3). Melanocytes in café-au-lait spots (CALMs) and Schwann cells in neurofibromas have disease-causing variants in both alleles of NF1 (4). Segmental NF1 is the result of mosaic disease-causing variants in NF1, and can be difficult to detect, as conventional genetic testing is performed on blood. Segmental NF1 may therefore be underdiagnosed, and testing of lesional tissues should be considered (4).
Clinical findings
Diagnosis of NF1 is based on the clinical criteria requiring the presence of at least 2 of the following major criteria: 6 or more CALMs (> 5 mm pre-puberty and > 15 mm post-puberty), 2 or more cutaneous neurofibromas, one plexiform neurofibroma, axillary or inguinal freckling, optic gliomas, 2 or more Lisch nodules, characteristic bony defects, or a first-degree relative with NF1 (5). Almost all patients develop CALMs in the first year of life. Axillary and inguinal freckling is the most specific sign of NF1 (Fig. 1). Cutaneous neurofibromas tend to present during puberty and continue to increase in both number and size throughout life (4). Juvenile xanthogranulomas (JXGs) have also frequently been reported in children with NF1, and it has even been proposed to add JXG as a minor diagnostic criterion in children (6). Naevus anaemicus can also be a diagnostic clue, especially useful in smaller children (7).
Fig. 1. Neurofibromatosis type 1.
Aside from the cutaneous manifestations, skeletal abnormalities occur in up to 20% of patients with NF1, especially males, and include osteopaenia, scoliosis, sphenoid wing dysplasia, congenital tibial dysplasia and pseudarthrosis (3). Patients with NF1 are also predisposed to cognitive impairment and learning difficulties (8).
Extracutaneous neoplasms
Approximately 15–20% of patients develop low-grade optic gliomas, and also have a 5-fold increased risk of glioblastomas (9). Patients with NF1 have a lifetime risk of 8–16% of developing malignant peripheral nerve sheath tumours (MPNST), which are a subtype of sarcoma. Most occur from pre-existing plexiform neurofibromas or non-dermal neurofibromas and have a poor prognosis (9). Gastrointestinal stromal tumours can develop anywhere along the gastrointestinal tract, and approximately 95% are asymptomatic (3). Children with NF1 and JXGs may have a higher risk of developing juvenile myelomonocytic leukaemia than children with NF1 alone; however, this has been debated (10).
Differential diagnosis
Legius syndrome is a differential diagnosis to NF1 and presents with multiple CALMs and skinfold freckling, but without an increased risk of cancer. Legius syndrome is associated with disease-causing variants in SPRED1 (11).
Management and treatment
Patients with NF1 should be managed by a multidisciplinary approach. Clinical evaluation by a NF1 specialist is recommended every 1–3 years depending on phenotype. Annual examinations can be performed by a primary care physician, dermatologist or paediatrician (9). Treatment of symptomatic neurofibromas consists of surgical excision, although small lesions can be managed with CO2 laser, radiofrequency, electrodessication or ablation (12). To date, there is no topical or systemic medical treatment recommended for cutaneous neurofibromas. Clinical trials of MEK inhibitors have shown promising results in treating both peripheral nerve sheath tumours and optic gliomas (9, 13).
Neurofibromatosis type 2 (NF2) is far less frequent than NF1, and although the disease is defined as “neurofibromatosis”, neurofibromas are, in fact, relatively infrequent in NF2 (14). NF2 was first described in 1822 by Wishart (15), and in 1916 Cushing described bilateral tumours on the eighth cranial nerve as a part of NF1 (16). Cushing’s description is largely responsible for the confusion between NF1 and NF2, and it was not until 1987 that NF2 was recognized as a separate disorder (17).
Molecular genetics and pathophysiology
NF2 is an autosomal dominant disorder with a prevalence of 1 in 25,000 live births. Penetrance is almost 100% by the age of 60 years, although the expressivity is highly variable (14). NF2 is caused by disease-causing variants in the tumour suppressor gene NF2, which is located on chromosome 22q12.2 and encodes for merlin, also known as schwannomin. Subsequently, tumours develop from cells that lose function of wild-type NF2 allele in susceptible target organs, such as the nervous system, eyes and skin (14).
Clinical findings
Diagnosis of NF2 was previously based on the Manchester criteria (18). However, Baser et al. (19) have developed an improved set of diagnostic criteria based on current understanding of the natural history and genetic characteristics of NF2 (20). According to the Baser criteria, a diagnosis of definite NF2 is established if the patient achieves a score of at least 6 points (19). Cutaneous features are much more subtle in NF2 than in NF1. A total of 59–68% of patients present with skin tumours, including skin plaques, subcutaneous tumours and intradermal tumours (14), but only 10% present with more than 10 skin tumours. The majority of the tumours are histologically schwannomas (21). Intracutaneous plaque-like lesions with hyperpigmentation and hypertrichosis are most frequent. More deep-seated subcutaneous nodular tumours often occur along major peripheral nerves and are palpable (21). CALMs present in up to 48% of patients with NF2, but are often solitary (14).
Extracutaneous neoplasms and symptoms
NF2 is characterized by the development of vestibular schwannomas (VS): 90–95% of patients develop VS, and they generally present with hearing loss and/or accompanying tinnitus, dizziness and imbalance. Although VS are benign, they are a substantial cause of morbidity due to their location. Other tumours, such as intracranial meningiomas, occur in 45–58% of patients with NF2, spinal meningiomas in 20% and ependymomas in 18–53%, but the last type cause symptoms in less than 20% of patients (14).
Management and treatment
Patients diagnosed with NF2 should be managed by a multidisciplinary team and followed up with annual neurological and audiological examinations. Ophthalmological evaluation should be performed in selected patients with visual impairment or facial weakness (20). Pre-symptomatic children with an affected parent should be screened: cranial/spinal magnetic resonance imaging (MRI) and ocular examinations are recommended, because children often present with cataract or retinal hamartomas as the first sign of NF2. Screening in alternative years below the age of 20 years, and every 3–5 years after the age of 20 years is acceptable (22). Complete surgical resection of the VS is curative, but the timing is controversial (20). Even with improvements in microsurgery and with use of radiation therapy, the majority of individuals lose their hearing completely (14, 21). As an alternative, or in addition to tumour removal, stereotactic irradiation and/or chemotherapy can be used to delay progression, but it is thought to increase the risk of secondary malignancies. At present, there is no effective chemotherapy for treatment of NF2-related tumours. Recently, several promising pharmacological strategies have been developed following the studies on merlin pathway (20). Different therapeutic targets have been proposed and the vascular endothelial growth factor (VEGF) inhibitor bevacizumab has shown the most promising results in treating VS (23).
The characteristic skin lesions of tuberous sclerosis complex (TSC) were reported for the first time in 1835 by John James Pringle (24). In 1862, von Recklinghausen first reported cerebral involvement in TSC. Description of the cerebral pathology is, nevertheless, credited to Désiré Magloire Bourneville, who proposed the term ” sclérose tubéreuse des circonvolutions cérébrales” to characterize the islets of sclerosis he found in the cortical gyri of 2 patients with TSC (25). It was not until the beginning of the 20th century that a more complete clinical picture of TSC was described. Today TSC is known to be a syndrome with benign tumours or hamartomas in many organs, but particularly skin, brain, eye, kidneys and heart (26).
Molecular genetics and pathophysiology
TSC is an autosomal dominant disorder with a birth rate of approximately 1 in 6,000 live births (26). TSC is caused by disease-causing variants in either TSC1, located on chromosome 9q34 encoding hamartin, or TSC2, located on chromosome 16p13.3 encoding tuberin. These gene products inhibit the mammalian-target-of-rapamycin (mTOR) pathway. mTOR detects signals of nutrient availability, hypoxia or growth factor stimulation and plays an important role in cortical development and growth control (26, 27). If conventional genetic testing is normal, intronic mutations or mosaicism should be considered (28). Two-thirds of patients with TSC result from sporadic disease-causing variants and many of these cases represent mosaicism (29). Next generation sequencing can identify most mosaic variants. Patients with mosaic TSC tend to have a lower overall severity, but no distinctive clinical features (30).
Clinical findings
Diagnosis is made by the presence of 2 major features or 1 major and ≥ 2 minor features. Major features include: ≥ 3 hypomelanotic macules (at least 5-mm diameter), ≥ 3 angiofibromas or fibrous cephalic plaque, ≥ 2 ungual fibromas, shagreen patch, multiple retinal hamartomas, cortical dysplasia, subependymal nodules, subependymal giant cell astrocytoma, cardiac rhabdomyoma, lymphangioleiomyomatosis (LAM) and ≥ 2 angiomyolipomas. Minor features include: “confetti” skin lesions, > 3 dental enamel pits, ≥ 2 intraoral fibromas, retinal achromic patch, multiple renal cysts and non-renal hamartomas (31). The manifestations can vary greatly, but cutaneous manifestations occur in more than 90% of patients. Angiofibromas are the most common cutaneous manifestation, being present in 75% of patients with TSC (Fig. 2) (32). Nipple angiofibromas can be found in up to 20% of patients (33). Other skin lesions consist of hypomelanotic macules (common in infants), ungual or gingival fibromas, and thickened, firm areas of subcutaneous tissue, often on the lower back, buttocks, torso (shagreen patches), forehead and face (fibrous plaques) (26, 27). More than 90% of patients with TSC develop central nervous system complications, including epilepsy, which can be complicated and a treatment challenge. TSC is also associated with a range of neuropsychiatric manifestations, such as behavioural, intellectual and psychosocial, termed tuberous-sclerosis-associated neuropsychiatric disorders (TAND) (32).
Fig. 2. Tuberous sclerosis. Written permission was given to publish this photograph.
Extracutaneous neoplasms
Approximately 60% of patients with TSC present with cardiac rhabdomyomas, which makes these the most common neoplasm in TSC (32). Although patients often have multiple tumours, they are rarely symptomatic and usually recede over time. Renal angiomyolipomas are found in approximately 80% of adult patients and kidney complications are the most frequent TSC-related cause of death (26, 32). Approximately 50% of patients develop renal cysts (32) and 2–3% of patients develop renal cell carcinomas (27). Retinal hamartomas are found in approximately 50% of patients with TSC and are also found in newborns (27). Although pulmonary LAM presents late compared with other symptoms, it is associated with high morbidity and mortality. Approximately 10–30% of patients with TSC, mostly women, present with LAM (34). A number of cerebral lesions are found in patients with TSC, including cortical tubers, subependymal nodules and subependymal giant cell astrocytoma (27, 32).
Management and treatment
Patients diagnosed with TSC should have a baseline status performed, which includes: dermatological clinical evaluation, ophthalmological screening, neurological examination, electroencephalography, cerebral MRI, electrocardiogram, echocardiogram, renal ultrasound and MRI of the abdomen, pulmonary function testing, and glomerular filtration rate (35). Ongoing periodic surveillance is needed after diagnosis for optimal care and prevention of complications. Surveillance protocols are described in Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference (35). When treating TSC-associated cutaneous disorders both pharmacological and non-pharmacological approaches can be effective. Facial angiofibromas may become symptomatic and a cosmetic problem, but topical sirolimus has shown to be both efficacious and well tolerated in long-term treatments (> 52 weeks) (36). Before the advent of mTOR inhibitors, non-pharmacological therapies, such as pulsed-dye laser, ablative lasers and a range of surgical approaches, were the mainstay and, in some cases, are still the treatment of choice (26, 37).
Gardner syndrome (GS) belongs to the familial adenomatous polyposis syndrome (FAP). Eldin Gardner described the syndrome in 1951 (38), where he reported a significant correlation between external osseous and cystic tumours and polyposis of the colon, which had a high malignant potential. In 1953, Gardner & Richards (39) described GS as hereditary colonic polyposis associated with osteomas and multiple soft tissue tumours. The combination of intestinal polyposis and extraintestinal manifestations, such as osteomas, dental abnormalities and benign soft tissue tumours, was termed GS in honour of Eldin Gardner, but it is now clear, that these patients should be classified within the spectrum of FAP (40).
Molecular genetics and pathophysiology
FAP is an autosomal dominant inherited disorder with an incidence reported from 1:6,850 to 1:23,700 live births (41). The syndrome is estimated to account for approximately 20–40% of patients with FAP (42). Other variants of FAP are attenuated FAP (AFAP), in which individuals are more mildly affected, and gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS) (40). Both GS, FAP and other variants are caused by disease-causing variants in the tumour suppressor gene adenomatous polyposis coli (APC) located on chromosome 5q21-22 (42). This gene encodes a protein that inhibits the Wnt signalling pathway by downregulating beta-catenins activity. The loss of function in APC leads to accumulation of beta-catenins, which bind to several transcription factors, altering the expression of genes involved in proliferation, differentiation, migration and apoptosis.
Clinical findings
A clinical diagnosis of classical FAP is based on the presence of > 100 colorectal adenomas. When a patient presents with multiple or familial cysts, located on unusual sites (such as the limbs) or with unusual histological features (mixture of epidermoid, trichilemmal cysts and/or pilomatricomas), GS/FAP should be considered (42). Skin findings also include “Gardner fibromas” on the trunk, which consist histologically of sheets of collagen bundles with interspersed bland fibroblasts, and nuchal fibromas that may cause swelling of the neck (42). Angiofibromas, lipomas, leiomyomas and neurofibromas are rarely seen. Other findings include congenital hypertrophy of the retinal pigment epithelium (CHRPE), which is the earliest and most common potential extraintestinal manifestation of FAP and affects up to 80% of the patients, although this finding alone is not diagnostic (43).
Extracutaneous neoplasms
Intestinal polyposis and colorectal adenocarcinomas are hallmarks of GS. Already in the second or 3rd decade of life patients develop hundreds to thousands of adenomatous polyps, and colorectal cancer occurs in nearly 100% of patients, if untreated, with a mean age of 39 years at cancer diagnosis (44). Adenomatous polyps of the duodenum are observed in 50–90% of individuals with FAP, and the risk of duodenal cancer is 4–12% (40). Gastric polyps are found in most patients with FAP and 10–15% develop desmoid tumours with significant morbidity and mortality (45). Other tumours associated with GS/FAP include osteoma (20%), thyroid cancer (2.6 %), adrenal cancer, medulloblastoma, ependymoma, astrocytoma, hepatoblastoma (1.6%), gastric cancer (0.6%) and pancreatic cancer (40, 42, 44, 46).
Management and treatment
Skin manifestations are treated according to their specific type. The recommendations for patients with FAP include: annual or biannual colonoscopy, starting at the age of 10–12 years or whenever there are suggestive symptoms (chronic diarrhoea, rectal bleeding or abdominal pain) (40, 44); oesophagogastroduodenoscopy every 6 months to 4 years starting at age 20–30 years, depending on duodenal adenoma burden; supplemental CT or MRI for visualization of the small bowel should be considered; annual physical examination for extracolonic manifestations and palpation of the thyroid and abdomen starting in the late teenage years; annual thyroid screening with ultrasound, and, finally, screening for hepatoblastoma by liver ultrasound and measurement of serum alpha-fetoprotein concentration until the age of 5 years should be considered. Preventive colectomy is advised at the end of the second or third decade of life for all carriers of disease-causing variants (40). Different drugs, such as sulindac, erlotinib, tamoxifen, doxorubicin-dacarbazine and curcumin have been studied without consistent recommendations (40, 42).
It appears likely that a pair of identical twins with dark pigment spots on their lips and mucosa, described by Connor in 1895, were the first patients described in the literature with Peutz-Jeghers syndrome (PJS) (47). In 1921, Peutz was the first to describe PJS as a familial syndrome (48). Jeghers defined it as a distinct entity in 1949, and the syndrome was subsequently named “Peutz-Jeghers syndrome” (49, 50).
Molecular genetics and pathophysiology
PJS is an autosomal dominant disorder of unknown prevalence, with an estimated incidence of 1 in 50,000–200,000 people (50). PJS is caused by disease-causing variants in the serine/threonine kinase 11 gene (STK11), previously called LKB1, which is located on chromosome 19p13. STK11 is regarded as a tumour suppressor gene affecting the mTOR pathway and thereby regulating central cell processes, such as cell metabolism, apoptosis, polarity and proliferation.
Clinical findings
In a single individual, a clinical diagnosis may be made when any one of the following criteria is present: 2 or more histologically confirmed PJS polyps; any number of PJS polyps detected in one individual who has a family history of PJS in a close relative(s); characteristic mucocutaneous pigmentation in an individual who has a family history of PJS in a close relative(s) or any number of PJS polyps in an individual who also has characteristic mucocutaneous pigmentation (50). Of patients with PJS, 95% present with mucocutaneous lentigines located around the mouth, nostrils, perianal area, fingers, toes and the dorsal and volar aspects of hands and feet (Fig. 3). The lentigines tend to fade after puberty, but often persist in the buccal mucosa (50, 51).
Fig. 3. Peutz-Jeghers syndrome. Written permission was given to publish this photograph.
Extracutaneous neoplasms
A key feature of PJS is the occurrence of multiple polyps in the gastrointestinal tract. Approximately 60–90% of patients have polyps in the small bowel and 50–64% have colorectal polyps (50, 51). Some of the possible complications are bleeding (resulting in anaemia), intussusception and obstruction. PJS is associated with an elevated risk of cancer throughout the patient’s lifetime. This risk of cancer has been estimated to be: 39% for the colon and rectum, 11–36% for the pancreas, 29% for the stomach, 13% for the small intestine, 24–54% for the breasts, 21% for ovaries, 10–23% for the cervix, 9% for uterus carcinoma and Sertoli cell tumours of the testes and 7–17% for the lungs (52). Women with PJS also develop distinctive tumours, which include ovarian sex cord tumours with annular tubules, mucinous tumours of the ovary and well-differentiated adenocarcinomas of the cervix.
Management and treatment
All patients should be offered regular screenings. A consortium review group has recommended upper gastrointestinal endoscopy, video capsule endoscopy and colonoscopy to be performed from the age of 8 years (44). If polyps are found, the examinations should be repeated every 3 years. If no polyps are found, a second baseline examination should be performed at the age of 18 years and every 3 years thereafter. In addition, women should be recommended annual self-examination, starting at the age of 18 years, and annual breast MRI, and/or mammogram, starting at the age of 25 years. Regular testicular examinations should be offered to men, and ultrasound should be performed if abnormalities are palpated or if feminization occurs. Risk of lung cancer is increased, but no specific screening has been recommended; annual chest radiograph or chest CT in smokers should be considered. Treatment involves colonoscopic removal of polyps. Colectomy is sometimes necessary if colonoscopic management is difficult and especially if neoplastic changes are found. Chemoprevention to decrease polyp burden in PJS is being studied (COX-2 inhibitors, everolimus) (44). Many patients seek cosmetic treatment for the lentigines. There is no standard treatment, although there have been reports of successful improvement with intense pulse light or different types of laser (50, 51).
CYLD cutaneous syndrome (CCS) is a heritable condition characterized by multiple skin tumours that usually appear on the scalp (53). Historically, different descriptive names have been used, including Brooke Spiegler syndrome (BSS), multiple familial trichoepithelioma (MFT) and familial cylindromatosis (FC) (54). These syndromes are now recognized to constitute a clinical spectrum, and all phenotypes can present within CCS.
Molecular genetics and pathophysiology
CCS is a rare autosomal dominant disorder. The true prevalence of CCS is difficult to assess, because of the variability in presentation, but estimates suggest that approximately 1 in 100,000 people are carriers (53). A female preponderance has been described, due to an increased expressivity of CYLD mutations in women, who shows more severe phenotypes. CCS is caused by disease-causing variants in the tumour suppressor gene CYLD, which is located on chromosome 16q12-13. CYLD encodes an ubiquitin hydrolase, which negatively regulates the NF-κB pathway (53). If CYLD function is lost, the consequently increased NF-κB signalling may give tumour cells a survival advantage via proliferation and resistance to apoptosis. In addition, this ubiquitin hydrolase enzyme negatively regulates a range of signalling pathways including JNK, Wnt, TGFB1 and Notch, which are of importance in inflammation and cancer.
Clinical findings
The tumours include cylindromas, spiradenomas and trichoepitheliomas and are localized in the head and neck area (Fig. 4) (55). They can also be located on the torso, genitals and axillary skin (54). Some patients develop cylindromas with primary involvement of the scalp. In addition, patients often present with multiple small perinasal trichoepitheliomas. The tumours can appear as early as in childhood or puberty. They are often painful and tend to grow in number and size throughout life and can be cosmetically disfiguring (55). Formal criteria for CCS has not been established, but CCS should be suspected in an individual with multiple skin tumours (histologically confirmed cylindromas, spiradenomas, and/or trichoepitheliomas) and/or by identification of germline heterozygous disease-causing variants in CYLD by molecular genetic testing (56).
Fig. 4. CYLD cutaneous syndrome. Written permission was given to publish this photograph.
Extracutaneous neoplasms
Malignant transformation of cylindromas and spiradenomas in patients with CCS has been reported in approximately 5–10% of patients and shows a more aggressive pattern than in sporadic cases (55). A variant of trichoepithelioma, called trichoblastoma, can undergo malignant transformation (53, 55). Rare cases of salivary gland tumours have been reported, mostly affecting the parotid gland and usually after the age of 40 years (55). Rarely, pulmonary cylindromas develop in the large airways and may compromise breathing (54). The occurrence of cylindromas within the breast tissue is extremely rare (55).
Management and treatment
Due to the risk of malignant transformation, annual dermatological examinations are essential (54). Routine imaging is not currently recommended, unless malignant transformation is suspected. Because CCS is characterized by diffuse and numerous adnexal tumours, surgical excision may be difficult. Ideally, early excision is preferred to preserve as much normal scalp and skin as possible (54). Electrosurgery or laser ablation are other possibilities. Mohs micrographic surgery should be considered for recurrent tumours. Radiotherapy should be avoided. Multidisciplinary team management of tumours that have undergone malignant transformation is recommended.
Cowden syndrome (CS), now encompassed by the term PTEN hamartoma tumor syndrome, was originally described in 1963 by Lloyd & Dennis (57). It was named after the patient Rachel Cowden, in whom it was first reported. The patient presented with multiple hamartomas, unusual skin, facial and central nervous system findings combined with fibrocystic breast disease. Nine years later Weary described 5 more patients with similar findings (58).
Molecular genetics and pathophysiology
CS is an autosomal dominant syndrome with a Caucasian and female predominance, and affects approximately 1 in 200,000 live-born infants (59). CS is caused by disease-causing variants in the tumour suppressor gene PTEN (phosphatase and tensin homologue), located on chromosome 10q22-23. PTEN gene encodes a dual phosphatase protein, which negatively regulates the PI3K/Akt/mTOR pathway. Loss of function of this gene leads to increased cellular growth, migration, proliferation and survival (60).
Clinical findings
CS is diagnosed when an individual shows either 3 or more major criteria (including macrocephaly, Lhermitte-Duclos disease, or gastrointestinal hamartomas) or 2 major and 3 minor criteria (61). These criteria are summarized in Table I (61). The characteristic mucocutaneous features of CS are found in almost 100% of cases, with a mean age of onset of 22 years (60). The most common mucocutaneous lesions are facial trichilemmomas, papillomatous papules and acral keratoses. The facial trichilemmomas most commonly appear close to the hairline on the face and the neck. The papillomatous papules are most often found on the oral mucosa, the face and pressure points (hands and feet) (Fig. 5). The acral keratoses may resemble pits if they are small, or papules if they are larger (Fig. 5) (61).
Table I. Diagnostic criteria for Cowden syndrome (61)
Fig. 5. Cowden syndrome.
Extracutaneous neoplasms
Tan et al. (62) have published a large cohort of 368 patients with CS and have estimated the lifetime risk of different cancer types in this population: the risk of female breast cancer was 85%, for endometrial cancer 28%, for thyroid cancer 35%, for kidney cancer 34%, for colorectal cancer 9%, and for malignant melanoma 6%. The risks seem to be higher than previously reported, and the authors recommend considering colorectal and kidney cancers and melanoma as members of the cancer spectra arising from germline mutations of PTEN. More than 90% of patients with CS develop multiple polyps throughout the gastrointestinal tract with different histological presentations: hamartomas (the most frequent), hyperplastic polyps, ganglioneuromas, adenomas, inflammatory polyps, leiomyomas, lipomas and lymphoid polyps (63).
Management and treatment
Women are recommended a clinical breast examination from the age of 25 years and annual mammography or MRI from the age of 30 years (64). Prophylactic mastectomy is an option to consider. All patients should undergo annual thyroid ultrasound examinations starting at the time of diagnosis, including in childhood. Annual dermatological examination may be indicated. Renal ultrasound, starting at age 40 years and every 1–2 years, should be considered. Colonoscopy should start at age 35 years and every 5 years, unless symptoms indicate earlier examination. If the patient has a close relative with colon cancer before the age of 40 years, then colonoscopy is recommended 5–10 years before the earliest colon cancer in the family. Screening for endometrial cancer via endometrial biopsy every first or second year can be considered. Facial papules can be treated if cosmetically disfiguring, although patients will continue to develop new papules throughout life. Treatment options include topical 5-fluorouracil, surgical removal and CO2 laser (60). Future treatments may focus on inhibition of the PI3K/Akt/mTOR pathway: non-steroidal anti-inflammatory drugs (NSAIDs) have been shown to downregulate Akt in PTEN (60). Furthermore, the effect of using a mTOR inhibitor, such as rapamycin, may represent a suitable option for chemoprevention and treatment of the defective PTEN function.
Carney complex (CNC) was first described by Carney in 1985 (65) as “the complex of myxomas, speckled pigmentation and endocrine over-activity”. The syndrome was later designated as CNC by Bain (66), and it was suggested that the majority of patients, who were previously diagnosed as LAMB (lentigines, atrial myxoma, mucocutaneous myxoma, blue naevi) or NAME (naevi, atrial, myxoma, myxoid neurofibroma, ephelide) could be considered as having CNC (67).
Molecular genetics and pathophysiology
CNC is an extremely rare autosomal dominant disorder with an unknown prevalence. Centres in the USA and France have collectively reported more than 750 cases in the literature (68). CNC is caused by disease-causing variants in the PRKAR1A gene, which is located on chromosome 17q22-24. PRKAR1A is coding for the regulatory subunit type I alpha of the protein kinase A enzyme. It has been implicated in endocrine tumorigenesis and is considered a tumour suppressor gene (68, 69).
Clinical findings
The diagnosis is based on 2 major criteria or 1 major criterion plus 1 supplemental criterion. Major criteria are: lentiginosis, myxoma (cutaneous and mucosal) or cardiac myxoma, breast myxomatosis or fat-suppressed MRI findings suggestive of this diagnosis, primary pigmented nodular disease (PPNAD) or paradoxical positive response of urinary glucocorticosteroids to dexamethasone administration during Liddle’s test, acromegaly due to growth hormone-producing adenoma, testicular tumours (large-cell calcifying sertoli cell tumour) or characteristic calcification on testicular ultrasonography, thyroid carcinoma (at any age) or multiple hypoechoic nodules on thyroid ultrasound in a prepubertal child, psammomatous melanotic schwannomas, blue naevus or multiple epithelioid blue naevus, multiple breast ductal adenoma and osteochondromyxoma. Supplemental criteria are: affected first-degree relative, activating pathogenic variants of PRKACA and PRKACB and inactivating pathogenic variants of the PRKAR1A gene (68). The cutaneous manifestations are most often mucocutaneous lentigines, cutaneous or mucosal myxomas and blue naevi. Lentigines occur in approximately 75% of patients and may be located everywhere on the body, but they are most common on the face, lips, conjunctiva, inner and outer canthi, and vaginal and penile mucosa (Fig. 6). Lentigines are one of the early manifestations (peripubertal), but fade after the age of 40 (68–70) years. Cutaneous myxomas occur in approximately 30–50% of patients, and the distribution includes eyelids, ears, breasts and nipples (68). They usually present during the teenage years. Other associated skin manifestations include intense freckling, CALMs, multiple skin tags or other skin lesions, such as lipomas and angiofibromas (67, 68). Of the patients diagnosed with CNC 25–60% develop PPNAD, and adrenocorticoid hormone-independent Cushing syndrome due to PPNAD is observed in 25–30% (69).
Fig. 6. Carney complex. Written permission was given to publish this photograph.
Extracutaneous neoplasms
Patients with CNC have an increased risk of internal malignancies and endocrine disturbances (67–70). Thyroid cystic or multinodular disease can be found in up to 75% of patients and is generally caused by follicular adenoma, although up to 10% will result in a thyroid carcinoma (68, 69). Pituitary adenomas can also occur and cause acromegaly, but they usually appear in the third decade of life (70). Unique for CNC is the psammomatous melanotic schwannoma (PMS), which can occur in both the central and peripheral nervous system (67). Breast myxomas and ductal adenomas present after puberty in females with CNC (68). Both males and females may develop myxomas of the nipple at any age. Prolactinomas have rarely been described.
Management and treatment
Patients with CNC should undergo the following evaluations at least yearly: echocardiogram beginning in infancy (if cardiac myxoma is identified, biannually), skin evaluation by a dermatologist, blood testing for growth hormone, prolactin and insulin-like growth factor hormone 1 (IGF-1), urinary free cortisol, thyroid gland examination, and imaging techniques for detecting PPNAD and PMS. In males, annual testicular ultrasound examinations and, in females, annual transabdominal ultrasound of ovaries are recommended. In prepubertal children growth rate and pubertal stages must be closely monitored (68). The treatment of Carney complex is directed toward the specific symptoms that appear in each individual. Cardiac myxomas should be removed surgically, but the risk of recurrence is high (68, 70). There is no standard treatment for the cutaneous manifestations.
In 1975, Hornstein & Knickerberg (71) reported 2 siblings with perifollicular fibromas alongside a family history with similar skin lesions. One of the siblings had multiple colon polyps and subsequently developed colon cancer. In 1977, Birt, Hogg & Dubé (72) provided the first description of the autosomal dominant inheritance of this disorder and diagnosed the characteristic skin lesions as fibrofolliculomas.
Molecular genetics and pathophysiology
Birt Hogg Dubé Syndrome (BHDS) is an autosomal dominant disorder with an unknown prevalence; however, more than 600 families have been reported (73). BHDS is caused by disease-causing variants in the FLCN gene located on chromosome 17p11.2, which codes for folliculin. The gene appears to have a role in regulating cellular metabolism through AMPK and mTOR signalling pathways (74–76).
Clinical findings
Diagnosis is based on the criteria proposed by the European BHDS consortium: a patient has BHDS if one of the following is present: the patient has a pathogenic FLCN mutation; the patient has >4 fibrofolliculomas or trichodiscomas (Fig. 7), at least one histologically confirmed, of adult onset; or if 2 of the 3 following manifestations are present: (i) multiple bilateral lung cysts with a basal predominance and no other apparent cause, (ii) a first-degree relative with BHDS, (iii) early debut (< 50 years) of renal tumours or the presence of multiple bilateral renal tumours, or (iv) renal tumours of the chromophobe/oncocytotic type (75). Most often skin tumours occur in the third or fourth decade of life (75). There have been several cases of malignant melanoma reported in patients with BDHS (77). Of patients with BDHS, 67–90% develop lung cysts, which increases the risk of pneumothorax, which is often the first symptom in patients with BHDS (75). BHDS should be considered in patients with spontaneous pneumothorax or cystic lung disease without any obvious explanation (74–76).
Fig. 7. Birt-Hogg Dube.
Extracutaneous neoplasms
Approximately 30% of patients with BHDS will develop renal tumours, although the malignancy potential varies (76). In contrast to sporadic renal cell cancer, in patients with BHDS tumours tend to be multifocal and/or bilateral. Sattler et al. (78) recently published a large cohort of 178 patients/50 families with BHDS: 19.1% developed renal cell cancer and most kidney malignancies (64%) occurred after the age of 50 years. The hybrid chromophobe-oncocytoma cancer subtype was previously reported to have a 50% frequency in BHDS renal cell cancer, but in this cohort was 19.1%. The clear cell renal cell cancer was the most common histological subtype in the sample (47.6%). In earlier reports clear cell renal cell cancer accounted for only approximately 9% of all renal tumours in patients with BHDS (78). There have also been reports of parathyroid adenoma, lipomas, parotid oncocytoma and colonic polyposis, although many of these reports seem to be coincidental (74–76).
Management and treatment
The following examinations are recommended at diagnosis or when the patient is at least 20 years old: pulmonary function test every second year (if no or very mild symptoms) or otherwise high-resolution computed tomography (CT); MRI scanning of the kidneys, starting at the age of 20 years and every second year from the age of 24 or 25 years and referral to the dermatologist for a baseline evaluation (79). In patients with renal tumours, nephron-sparing surgery is advised due to the high number of patients developing bilateral or multiple renal tumours (74, 76). Lung cysts are not malignant, but can limit the patients in their choice of profession and hobbies. Pleurodesis or similar treatments should be considered in order to reduce the risk of recurrence (76, 79). Patients with multiple lesions located on the face may seek treatment for cosmetic purposes, but there is no standard treatment for the skin lesions of BDHS: surgical and laser treatments may lead to improvement, but often only temporary (75, 76, 79).
Hereditary leiomyomatosis and renal cell cancer (HLRCC), formerly known as Reed’s syndrome, was first described by Blum & Jean in 1954 (80). In 1973, Reed delineated the members of 2 families, whose successive generations developed cutaneous or uterine leiomyomas (81). The syndrome was renamed HLRCC in 2001, when the condition was linked to an increased susceptibility to certain types of renal cell cancer (82).
Molecular genetics and pathophysiology
HLRCC is an autosomal dominant inherited disorder with an unknown prevalence, but 200–300 families have been reported (83). HLRCC is caused by disease-causing variants in the tumour suppressor gene “fumarate hydratase” (FH), also called fumarase, which is located on chromosome 1q42.3-43 (84). FH encodes the enzyme catalysing the conversion from fumarate to malate in the Kreb’s cycle. The mechanism of tumorigenesis in fumarase-deficient cells is poorly understood, although hypoxia-inducible factors are hypothesized to play a role, with subsequent transcription of target genes involved in angiogenesis, cell proliferation, cell survival and invasion (85).
Clinical findings
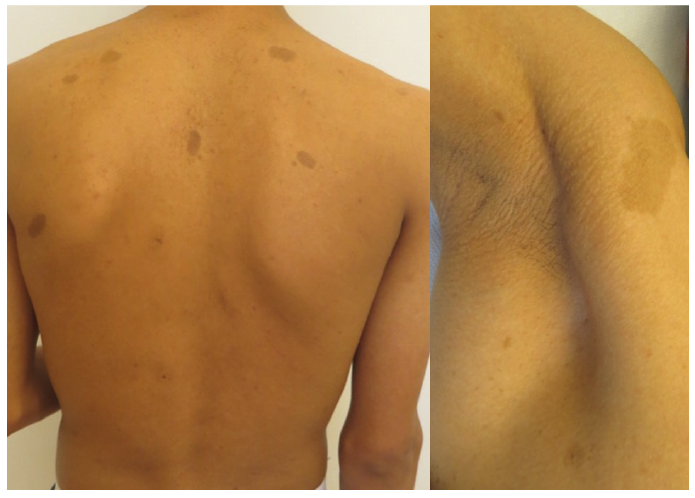
According to the revised diagnostic criteria, the diagnosis is definitive in case of detection of pathogenic germline fumarate hydratase mutation (83). The diagnostic criteria for HLRCC proposed by Hansen et al. (83) include one major criterion and 3 minor criteria. The major criteria “highly suggestive of HLRCC” are: multiple cutaneous leiomyomas with at least one histologically confirmed or family disposition to HLRCC and at least one minor criterion. The minor criteria are: single solitary histologically confirmed cutaneous leiomyoma, multiple severely symptomatic uterine leiomyomas with onset < 40 years, or type 2 papillary renal cell carcinoma with onset < 40 years. Having a first-degree family member who meets at least 1 of the above-mentioned criteria is also suspicious of HLRCC. HLRCC is characterized by cutaneous leiomyomas (CLM), which are often the first manifestation of the syndrome, as the mean age of onset is 25 years (84). CLM are smooth-surfaced, skin-coloured or erythematous solitary or multiple papules or nodules (Fig. 8) found in up to 100% of HLRCC families (83). Most CLM occur on the trunk and extremities, and occasionally on the face. Pseudo-Darier sign may be present. CLM are painful in approximately 90% of patients, and the pain may be spontaneous or induced by cold, stress, pressure or emotions (84). They increase in size and number throughout life. Malignant transformation into leiomyosarcoma is very rare.
Fig. 8. Hereditary leiomyomatosis and renal cell cancer.
Extracutaneous neoplasms
Uterine leiomyomas are present in 73–100% of females with HLRCC (84) and tend to be numerous and large and cause irregular menstruation and pain. Renal cell cancer is diagnosed in approximately 14–62% of HLRCC families, and in 16–24% of individuals with HLRCC (83). Renal cell carcinomas in patients with HLRCC are usually unilateral, solitary and aggressive, and display a spectrum of architectural patterns.
Management and treatment
There is a lack of consensus regarding a surveillance programme for patients with HLRCC. Dermatological examinations should be considered every year or every second year, starting from the onset of CLM, in order to evaluate changes suggestive of leiomyosarcoma (83, 84). Annual gynaecological examinations with ultrasound, starting at the age of 20 years, are recommended, and annual MRI of the kidneys is advised from the age of 8 or 10 years. CLM are benign, and therefore the need for treatment depends mainly on pain and cosmetic appearance. Surgical excision, electrodessication, cryosurgery and CO2 laser ablation are some of the surgical options. Topical lidocaine or capsaicin, botulinum toxin or triamcinolone acetonide injections or systemic treatments, such as nitroglycerine, nifedipine, gabapentin, phenoxybenzamine or doxazocin, may offer pain relief. Combination therapy may be useful. Uterine leiomyomas can be treated surgically (myomectomy or hysterectomy) or with gonadotropin-releasing hormone agonists and progesterone-releasing intrauterine devices. Due to the aggressiveness of renal cell carcinomas, surgical intervention is recommended even in small tumours.
The list of genodermatoses associated with extracutaneous malignancies is extensive. Schierbeck et al. (86) explored Gorlin-Goltz syndrome, Bazex-Dupré-Christol syndrome, xeroderma pigmentosum, Muir-Torre syndrome, Rothmund-Thomson syndrome, Bloom syndrome and familial atypical multiple mole melanoma syndrome.
Bazex-Dupré-Christol syndrome is characterized by multiple basal cell carcinomas (BCC), hypotrichosis, follicular atrophoderma and multiple milia, most commonly on the face (87). Epidermoid cancer of the lung, undifferentiated carcinoma of the bladder, prolymphocytic leukaemias and adenocarcinoma of the prostate have been reported in patients with this syndrome.
Xeroderma pigmentosum is characterized by extreme sensitivity to sunlight that results in sunburn, pigment changes and a greatly elevated incidence of skin cancers (88). The disorder is associated with a 10-fold higher risk of neurological tumours compared with the background population (89). There is also an increased frequency of cancer in the oral mucosa or the lungs and leukaemia.
Bloom syndrome is characterized by skin photosensitivity, CALMs, poikiloderma, hypopigmented lesions and early onset of BCC and squamous cell carcinoma (SCC) (86, 90). Patients with Bloom syndrome have an increased risk of lymphoma, acute myelogenous leukaemia, gastrointestinal cancer, breast cancer and cancer of the genitalia and urinary tract.
Rothmund-Thomson syndrome is characterized by poikiloderma, sparse scalp hair, sparse or absent eyelashes and/or eyebrows, juvenile cataracts, short stature, skeletal abnormalities, radial ray defects and premature ageing (91). The patients also have an increased risk of osteosarcoma and cutaneous neoplasms. Fibrosarcoma, verrucous carcinoma, acute myeloid leukaemia, non-Hodgkin’s lymphoma, parathyroid adenoma and gastric carcinoma have also been reported.
Muir-Torre syndrome is characterized by sebaceous tumours (epitheliomas, adenomas and carcinomas) and keratoacanthomas (42). Visceral tumours are multiple in 40–50% of patients with this syndrome; the most common is colorectal cancer. The list of other reported cancers is extensive, and includes urogenital, breast, upper gastrointestinal tract, lung, larynx, parotid gland and hematopoietic malignancies.
Gorlin-Goltz syndrome, also known as naevoid BCC syndrome, is characterized by multiple BCC, palmoplantar pits and jaw cysts (92). Of these patients, 2–5% may develop a brain tumour, most commonly medulloblastoma.
Familial atypical multiple mole melanoma syndrome is characterized by multiple cutaneous melanocytic naevi, usually more than 50, and an extremely high risk of developing malignant melanoma (86, 93). These patients have also a high risk of developing other internal malignancies, pancreas cancer being the most common.
Howell-Evans syndrome is characterized by focal thickening of the skin of the hands and feet and is associated with an extremely high lifetime risk (approximately 95% at the age of 65 years) of developing SCC of the oesophagus (94).
Ataxia telangiectasia patients present cerebellar degeneration, skin manifestations and immunodeficiency (95). Skin findings include telangiectasia on sun-exposed areas, premature greying of the hair, vitiligo and warts. These patients have also an increased risk of lymphoma and leukaemia; however, breast, liver, gastric and oesophagus cancer have been reported.
Fanconi anaemia is characterized by congenital abnormalities (skeletal malformations of the limbs, short stature and microcephaly among others), bone marrow malignancies, cutaneous pigmentation abnormalities and solid tumours (96). Skin lesions include CALMs and hypopigmentation. These patients have a highly increased risk of leukaemia and solid tumours, particularly of the head and neck, skin as well as gastrointestinal and genitourinary tracts.
Dyskeratosis congenita is characterized by a classic triad of dysplastic nails, reticular pigmentation of the upper chest and neck, and oral leukoplakia (97). The patients also have an increased risk of myelodysplastic syndrome, acute myelogenous leukaemia, SCC of the head/neck and anogenital cancer.
Werner syndrome is a subtype of progeria, characterized by the premature appearance of features associated with normal ageing (98). Early findings include greying and loss of the hair and skin changes, such as ulceration, hyperkeratosis, tight skin, age spots and subcutaneous atrophy. ‘’Bird-like’’ facies is typical. These patients have a high risk of developing cancer, particularly soft-tissue sarcomas, leukaemia, melanoma, meningioma, bone tumours and thyroid cancer.
Beckwith–Wiedemann syndrome is an overgrowth syndrome, in which the cutaneous findings include facial naevus flammeus, earlobe creases and posterior helical pits (99). The patients have an increased risk of Wilms tumour and hepatoblastoma, but also rhabdomyosarcoma, adrenocortical carcinoma and neuroblastoma.
Wiskott-Aldrich syndrome is a disorder of the haematopoietic cells. The skin manifestations include petechiae, purpura, recurrent skin infections and chronic eczema (100). These patients have an increased risk of lymphoma.
Nijmegen breakage syndrome is a rare syndrome characterized mainly by microcephaly, immunodeficiency and a very high rate of malignancies, predominantly of lymphoid origin (101). However, leukaemia, medulloblastoma and rhabdomyosarcoma have also been described. CALMs and vitiligo are the most frequently reported cutaneous findings.
Multiple endocrine neoplasia type 2 patients are predisposed to medullary thyroid carcinoma and pheochromocytoma (102). Skin findings include neuromas of the tongue, lips and eyelids and lichen amyloidosis.
Early detection of the typical signs of genodermatoses is important, because those lesions may be a sign of an underlying predisposition to extracutaneous neoplasms. Therefore, the dermatologist has a distinctive role in identifying inherited syndromes that could be related to malignancies. The identification of these syndromes plays, in addition, a significant role in the handling of patients and planning of personalized follow-up surveillance screenings. Therefore, early detection is also important in the clinical practice of oncologists, and may affect the clinical course of the syndrome.


 Click to show fullsize
Click to show fullsize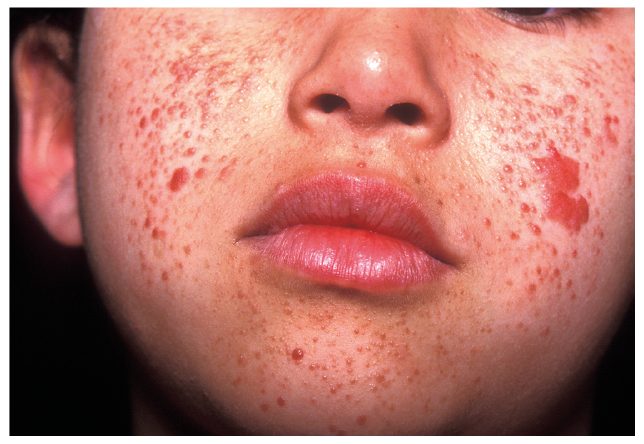 Click to show fullsize
Click to show fullsize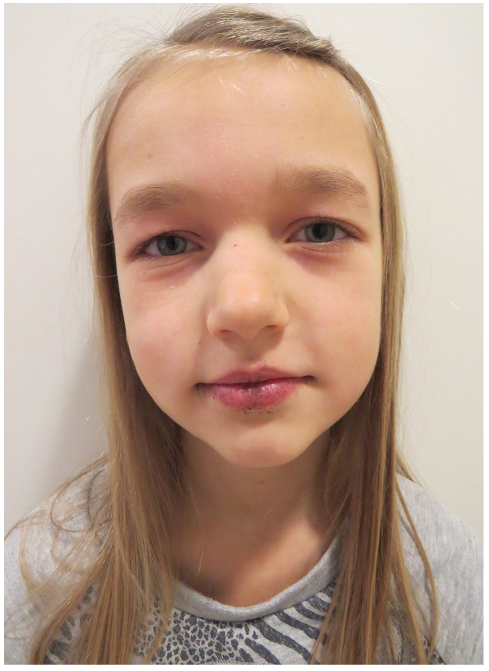 Click to show fullsize
Click to show fullsize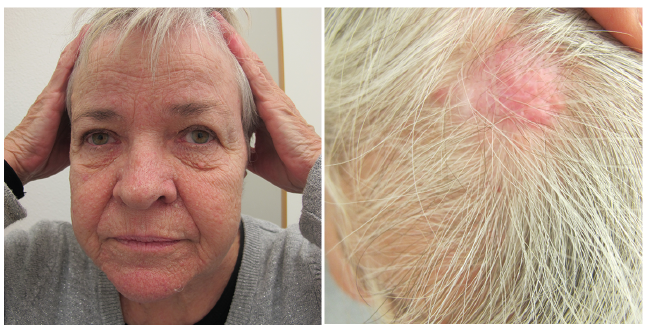 Click to show fullsize
Click to show fullsize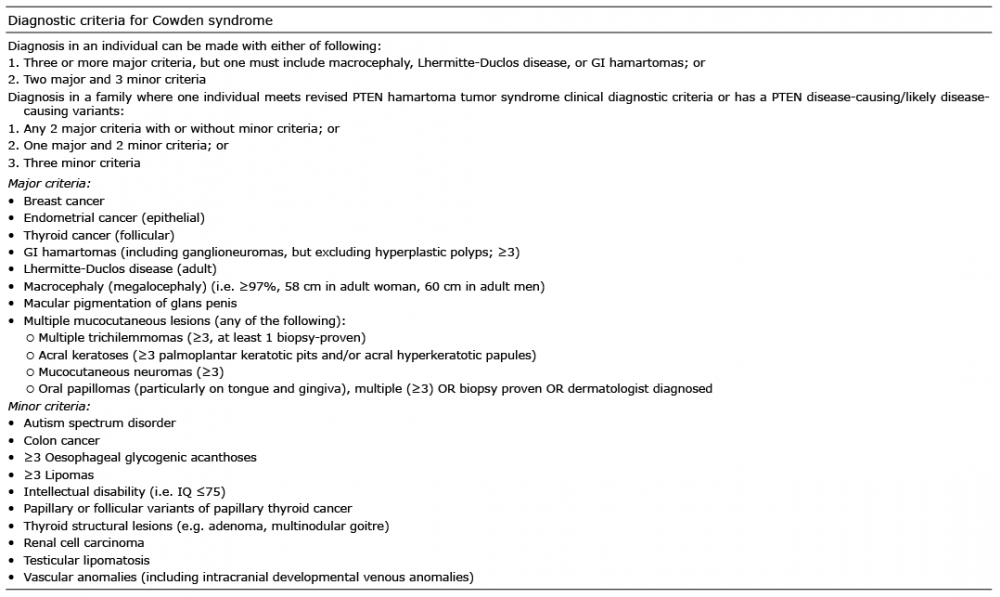 Click to show fullsize
Click to show fullsize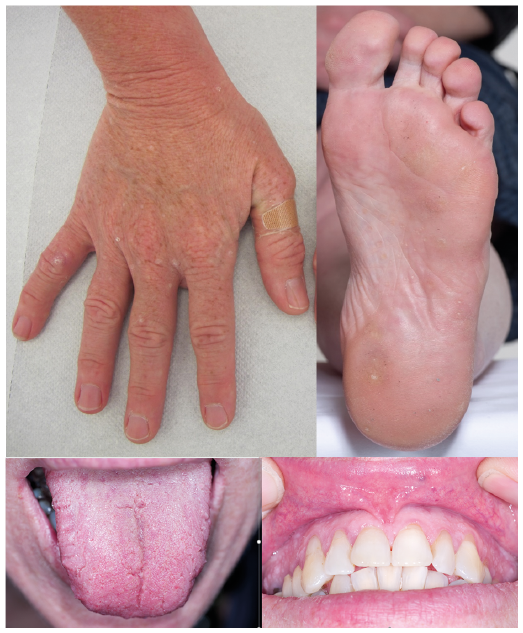 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize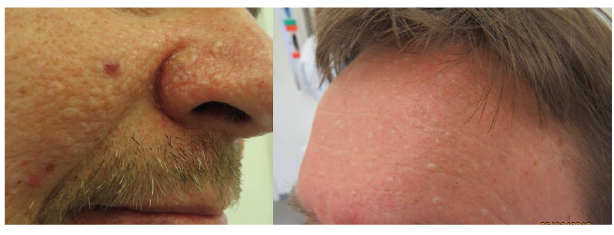 Click to show fullsize
Click to show fullsize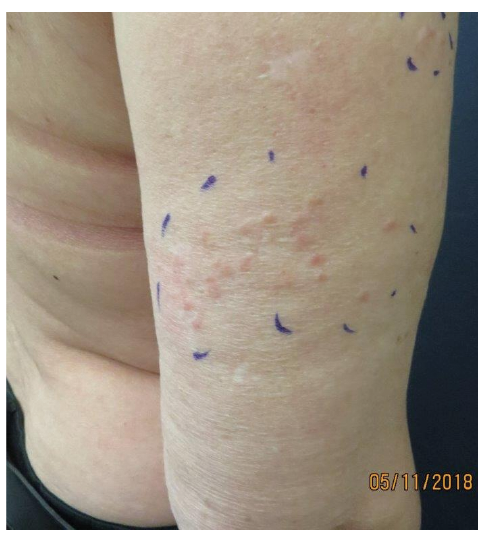 Click to show fullsize
Click to show fullsize