


1Department of Dermatology, National Cheng Kung University Hospital, 2School of Medicine, College of Medicine, 3International Center for Wound Repair and Regeneration (iWRR), National Cheng Kung University, Tainan, Taiwan, 4Department of Dermatology, Hokkaido University Graduate School of Medicine, Sapporo, Japan, 5Institute of Molecular Medicine, College of Medicine, 6Center for Genomic Medicine, Innovation Headquarters, 7Institute of Clinical Medicine, 8Center of Clinical Medicine, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan, 9St John’s Institute of Dermatology, King’s College London (Guy’s Campus), London, UK, 10Deparmtent of Genomic Medicine, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan. *E-mail: kylehsu@mail.ncku.edu.tw
#Co-first author.
Accepted Jul 6, 2021; E-pub ahead of print Jul 7, 2021
Acta Derm Venereol 2021; 101: adv00522.
doi: 10.2340/00015555-3874
Epidermolysis bullosa (EB) is a group of rare inherited blistering skin disorders, mostly caused by mutations in genes encoding structural proteins within the dermal-epidermal junction (DEJ) (1). Junctional EB (JEB) is one of the 4 classical subtypes characterized by blister formation within lamina lucida; inheritance is autosomal recessive underscored by bi-allelic mutations in LAMA3, LAMB3, LAMC2 (the laminin-332 genes), COL17A1 (type XVII collagen), ITGA6, ITGB4 or ITGA3 (α6, β4, α3 integrin subunits). Approximately 80% of patients with laminin-332-deficient JEB have mutations in LAMB3, encoding the β3 subunit of laminin-332, a heterotrimeric protein (α3, β3 and γ2 laminin polypeptides) at DEJ (2).
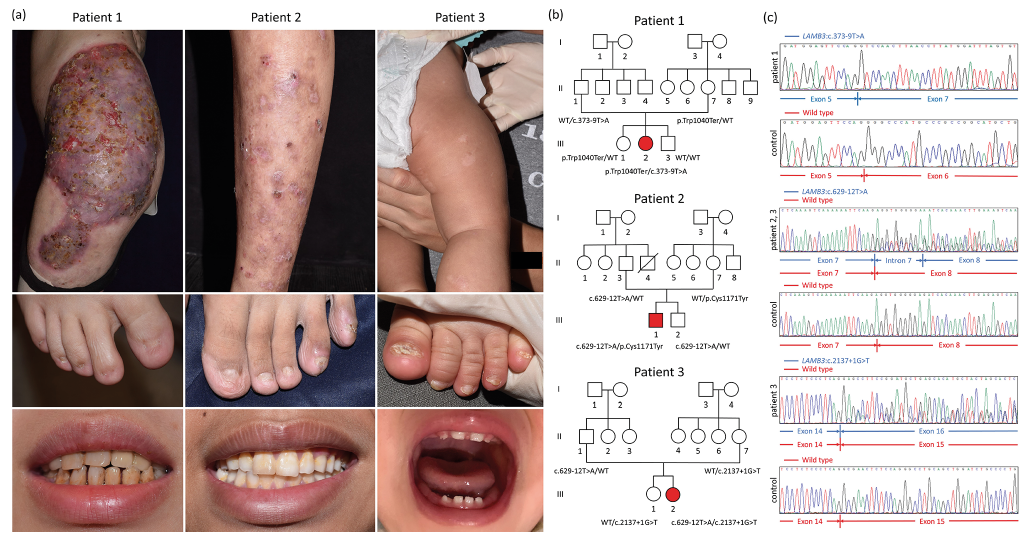
Although the laminin-332 molecular pathology in JEB may involve mutations in any of the LAMA3, LAMB3 or LAMC2 genes, it is unclear whether or how mutations in any of these genes affect the expression of the other 2 genes/proteins, and how this impacts on phenotype. We report 3 JEB patients presenting a variable degree of blistering, nail and teeth abnormalities (Fig. 1a) in whom we also explore the laminin-332 pathology in detail. Patient 1, a 29-year-old female, had recurrent erosions on thighs with persistent bacterial colonization. She also had nail dystrophy, corneal erosions, and dental enamel defects. Patient 2, a 16-year-old boy, had blisters, eczematous plaques on legs, nail dystrophy, but no visible enamel anomaly. Patient 3, an 18-month-old girl, had mild generalized blistering at birth with gradual spontaneous improvement. All her nails were dystrophic, with teeth being difficult to evaluate because only deciduous teeth were present.
Fig. 1. Clinical presentations and mutational analyses. (a) Diverse clinical manifestations of blisters, nail, and teeth abnormalities in the 3 patients with junctional epidermolysis bullosa. (b) All 3 cases carried compound heterozygous mutations in LAMB3 (NM_000228.3). Patient 1 carried c.373-9T>A (IVS6-9T>A) and c.3119G>A (p.Trp1040Ter); Patient 2 had c.629-12T>A (IVS8-12T>A) and c.3512G>A (p.Cys1171Tyr); Patient 3 harboured c.629-12T>A (IVS8-12T>A) and c.2137+1G>T (IVS15+1G>T). WT: wild type. Sanger sequencing results are shown in Fig. S1a. (c) Reverse-transcription PCR and Sanger sequencing confirmed the interference with LAMB3 mRNA splicing by all 3 splice-site mutations: c.373-9T>A caused the skipping of the entire exon 6; c.629-12T>A led to the retention of the first 10 bp in intron 7; c.2137+1G>T resulted in the skipping of exon 15.
Using whole-exome sequencing, compound heterozygous mutations in LAMB3 (NM_000228.3) were identified in all 3 cases: Patient 1: c.373-9T>A and c.3119G>A (p.Trp1040Ter); Patient 2: c.629-12T>A and c.3512G>A (p.Cys1171Tyr); Patient 3: c.629-12T>A and c.2137+1G>T (Fig. 1b, Fig. S1a). Among them, c.3119G>A, c.3512G>A and c.629-12T>A were reported (3). Reverse-transcription PCR and Sanger sequencing confirmed the interference with LAMB3 mRNA splicing by all 3 splice-site mutations: c.373-9T>A led to exon 6 skipping, causing in-frame deletion; c.629-12T>A caused retention of the first 10 bp in intron 7; and c.2137+1G>T resulted in exon 15 skipping, both of which caused frameshifts and downstream premature termination codons (Fig. 1c). Microfluidic electrophoresis demonstrated leakiness of all 3 splicing variants: mutant versus wild-type transcript level was 40:1 for c.373-9T>A, 1:3–1:7 for c.629-12T>A, and 3:1 for c.2137+1G>T (Fig. S1b). Immunofluorescence microscopy (IFM) demonstrated either absence or decreased staining for β3 and γ2 subunits, whereas α3 subunit immunoreactivity showed linear labelling (Fig. S2a). Quantitative PCR (Q-PCR) revealed significant decrease in LAMA3, LAMB3, LAMC2 transcripts (Fig. S2b). To investigate whether LAMA3 and LAMC2 reduction is reproducible in cultured keratinocytes, siRNA knockdown of LAMB3 in normal human epidermal keratinocytes was performed. Knockdown efficiency was greater than 90%, and LAMA3 transcripts were significantly reduced. LAMC2 was modestly reduced by 1 of the 2 LAMB3 siRNAs. The interference by LAMB3 knockdown was exclusive to laminin-332, since the transcription of other laminin genes expressed in keratinocytes and at DEJ, LAMA5, LAMB1, LAMC1, was unaffected (Fig. S2c). For further details, please see Appendix S1.
Gene expression, synthesis and the degradation of polypeptides that form complexes, such as laminins, are strictly regulated to maintain proportionality (4). For example, it was shown that mutations in LAMA3, LAMB3 or LAMC2 modulate transcription and expression of the other 2 subunits (5, 6). Current study investigated the impact of LAMB3 mutations on laminin-332 subunits at both protein and mRNA levels, revealing that nonsense-mediated mRNA decay hampers the abundance of LAMB3 transcripts (7) and further impedes the transcription of the 2 other genes. This implies that transcriptional activity may have mutual inter-dependence for these 3 genes, while further work is warranted to ascertain the significance and the detailed mechanism(s) therein (8).
In theory, protein expression should display some correlation with mRNA levels (5), although discrepancies are present in our study. Notably, despite significant decrease in LAMA3 transcripts, α3 subunit expression was nearly normal in all 3 patients. This may be explained by compensated translational efficiency, protein turnover (4) and α3 subunit also being expressed in another laminin isoform at DEJ, laminin-311 (9), which is expressed in both JEB skin and keratinocytes with LAMB3 mutations (10, 11). Besides, protein expression levels are presumed to be predictive of clinical severity and are related to the identified mutations (5). Indeed, immunoreactivity of laminin-332 antibody staining is often used for clinical diagnostics and prognostics (12). Here, however, further disparities were evident. Patient 2 has more severe phenotype than Patient 3, despite IFM showing greater intensity of GB3 staining, which was reported to be reflective of clinical severity in JEB (13). Both individuals have the same LAMB3 mutation, c.629-12T>A, but differ in their other mutations: Patient 2 has a missense mutation, p.Cys1171Tyr and Patient 3 has a splice-site mutation, c.2137+1G>T. This discordance between IFM and phenotypic severity is likely to relate to the leakiness of c.2137+1G>T (14) and the functional impact of the cysteine substitution: p.Cys1171Tyr in the β3 chain forms an interchain disulphide bond with the γ2 subunit and terminates the coiled-coil region of the heterotrimer (15), and thus p.Cys1171Tyr probably has a greater overall deleterious impact on functional laminin-332 levels. Collectively, current IFM studies reiterate published clinical experience that GB3, a primary antibody targeting a conformational epitope of γ2 subunit, is probably the best discriminator in the majority of JEB cases carrying LAMB3 mutations (13). GB3 immunolabelling was substantially reduced in Patient 1, but partially reduced in Patients 2 and 3, congruent with their degree of clinical severity. Notably, the relatively leaky splicing variants, c.629-12T>A and c.2137+1G>T, harboured by Patients 2 and 3 may, in part, explain the residual protein expression and milder phenotype (14).
In summary, current study demonstrates that LAMB3 mutations downregulate the other 2 laminin-332 encoding genes, LAMA3 and LAMC2. The discordances between transcription and protein expression alterations in the associated subunits of laminin-332 illustrate the complexity of the heterotrimer pathobiology and the clinical challenge in delineating accurate genotype-phenotype correlation in this group of patients.
This work was financially supported by the International Center for Wound Repair and Regeneration at National Cheng Kung University and the Ministry of Health and Welfare (rare disease prevention and control work and research projects) in Taiwan.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize