


1Division of Dermatology, Niigata University Graduate School of Medical and Dental Sciences, 1-757 Asahimachi-dori, Chuo-ku, Niigata 951-8510, 2Department of Dermatology, Nara Medical University School of Medicine, Kashihara and 3Department of Dermatology, Yamaguchi University Graduate School of Medicine, Ube, Japan. E-mail: shinkuma@naramed-u.ac.jp
Accepted Sep 2, 2021; Epub ahead of print Sep 7, 2021
Acta Derm Venereol 2021; 101: adv00547.
doi: 10.2340/00015555-3916
X-linked dominant chondrodysplasia punctata (XDCP; MIM 302960), also known as Conradi-Hunermann syndrome, is an X-linked dominant disorder caused by a mutation in the emopamil binding protein (EBP) gene (1, 2). It is characterized by skeletal, ophthalmological, and cutaneous manifestations. XDCP has been reported to present various manifestations, such as spontaneous resolution of ichthyosiform erythroderma following Blaschko’s lines, follicular atrophoderma, and cicatricial alopecia (3). While during the neonatal period, symptoms of XDCP are sometime severe, it gradually becomes mild in adulthood. We report here an adult female case of XDCP presenting pustular erythematous lesions caused by a novel heterozygous EBP mutation.
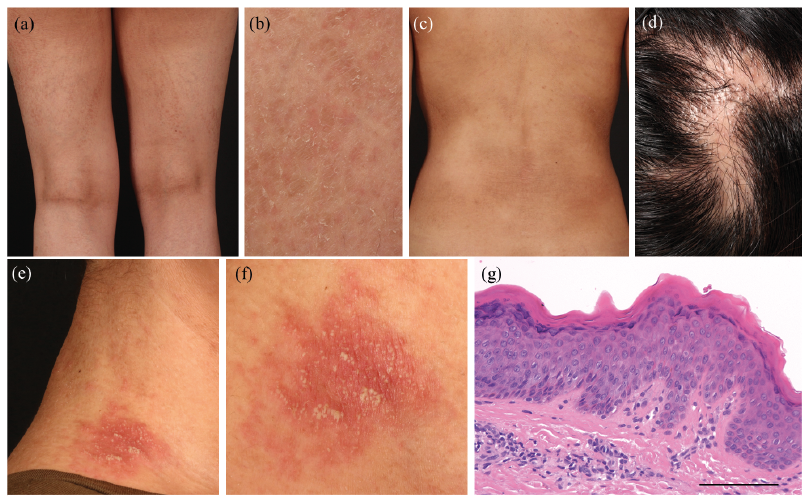
A 23-year-old woman with a lifelong history of xerosis sought our consultation. She was born to non-related healthy parents after an uncomplicated pregnancy. At birth, her skin was erythrodermic and exhibited desquamation without blistering. She did not have any siblings. Brownish linear streaky pigmentation with coarse scales along Blaschko’s lines (Fig. 1a, b) was initially observed on her entire body. Mild generalized dry skin was also noted unrelated to Blaschko’s line (Fig. 1c), and there were several linear areas of scarring alopecia on the scalp (Fig. 1d). Erythematous plaques with pustules were occasionally observed overlying the linear pigmentation along Blaschko’s lines on the neck and extremities; these pustules usually disappeared within a week (Fig. 1e, f). Skin biopsy of the linear coarse scales along Blaschko’s lines on the abdomen showed hyperkeratosis and a thickened granular layer without parakeratosis (Fig. 1g). She did not have ocular or auditory abnormalities, but her right leg was shorter than her left leg.
Fig. 1. Clinical manifestations of X-linked dominant chondrodysplasia punctata. (a, b) Linear pigmentation and ichthyotic scales along Blaschko’s lines on the thigh. (c) Linear pigmentation with coarse scales were noted on the trunk although xerosis was observed on the entire body regardless of Blaschko’s lines. (d) Cicatricial alopecia on the scalp. (e, f) Erythematous plaque with pustules was observed overlying the linear pigmentation along Blaschko’s lines on the neck. (g) Histopathological examination showing hyperkeratosis and thickened granular layer without parakeratosis. Haematoxylin and eosin (H&E) staining, original magnification ×200. Scale bar=100 μm.
We suspected an X-linked dominant disorder or somatic mosaicism, such as incontinentia pigmenti or systematized epidermal naevus, and performed whole exome sequence analysis of the DNA samples extracted from her whole blood after obtaining informed consent (under institutional approval and in adherence with the principles of the Declaration of Helsinki). Whole exome sequence analysis revealed a heterozygous 1-bp deletion (c.292delT) in exon 2 of EBP), leading to a frameshift and a subsequent premature termination codon (p.Ser98Leufs*40). Direct sequence analysis of the EBP gene revealed absence of the mutation in her parents (Fig. 2a, b). To the best of our knowledge, this mutation has not been previously reported and was not found in the gnomAD browser (https://gnomad.broadinstitute.org/) or human genetic variation database (http://www.hgvd.genome.med.kyoto-u.ac.jp/index.html). It was also confirmed that the deletion mutation was present in only 1 allele by direct sequencing after TA cloning (Fig. 2c). Reverse transcriptase PCR using RNA samples extracted from her blood samples showed markedly lower RNA expression levels in the patient compared with those in a healthy control, which confirmed that the mutation resulted in nonsense-mediated mRNA decay (Fig. 2d). We subsequently diagnosed the patient with XDCP based on her clinical and genetic characteristics. Treatment with topical corticosteroids and moisturizers resulted in a slight improvement of her condition.
Fig. 2. Mutation in the emopamil binding protein (EBP) gene. (a) Pedigree of the family. Affected individuals indicated in black and proband is indicated by an arrow. (b) Direct sequencing of EBP. The proband harbours a heterozygous mutation, c.292delT, in the EBP gene (red box). (c) Direct sequencing after TA cloning revealed a 1-bp deletion mutation on the mutant allele. (d) Reverse transcriptase-PCR of cDNA samples extracted from whole blood of the proband and healthy control. The primers were designed in exons 3–4 and 5. The EBP RNA expression levels in the patient were markedly lower than those in the healthy control.
XDCP is an X-linked dominant disorder caused by a mutation in the EBP gene located on the short arm of the X chromosome (Xp11.22-p11.23) (1, 2). In XDCP, approximately 70 different mutations have been described. The EBP gene encodes 3β-hydroxysterol-Δ8, Δ7-isomerase, which is involved in cholesterol synthesis. A mutation in EBP results in the accumulation of 2 cholesterol metabolites, 8(9)-cholestenol and 8-dehydrocholesterol, in tissues (4, 5). XDCP arises almost exclusively in females, and is usually intrauterine lethal in hemizygous males. Because patients with XDCP are born as collodion babies and have transient punctate calcifications in the epiphyseal regions during early infancy, it is relatively easy to diagnose XDCP in early childhood. Meanwhile, it is difficult to diagnose adult patients with XDCP, because ichthyosiform erythroderma improves with growth although a systematized linear pattern of atrophy showing mild ichthyosis often remains visible in adulthood (4). In fact, our case showed dry skin on the whole body, leading us to initially consider autosomal recessive congenital ichthyosis; however, the linear pigmentation with coarse scales along Blaschko’s lines contributed to the diagnosis of XDCP.
The clinical phenotype of XDCP is variable, owing to a random inactivation of the X-chromosome (4–6). Several reports have described the absence of a genotype-phenotype correlation (4, 5, 7–9). Typical cutaneous manifestations include spontaneous resolution of ichthyosiform erythroderma following Blaschko’s lines, follicular atrophoderma, and cicatricial alopecia (3). Notably, our patient occasionally presented with pustules within erythematous plaques. XDCP with pustules has been rarely reported; only one report of an adult female patient who developed erythematous papules and pustules on the trunk, inguinal regions, inner thighs, and lateral region of the neck along Blaschko’s lines that histopathologically resembled psoriasis has been documented (10). In the current study, skin biopsy from the pustular erythema was not conducted; therefore is difficult to compare the present patient to the case reported previously. It is notable that pustules may develop as a cutaneous manifestation of XDCP.
We report here an adult female with XDCP, who harboured a novel mutation in the EBP gene. Reverse transcriptase-PCR results revealed a marked decrease in the RNA expression level of EBP. Interestingly, the current patient occasionally presented with erythematous plaques with pustules. These observations underscore the importance of recognizing erythematous pustules as skin manifestations that can occur as a result of XDCP.
The patient provided consent for publication of this report.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize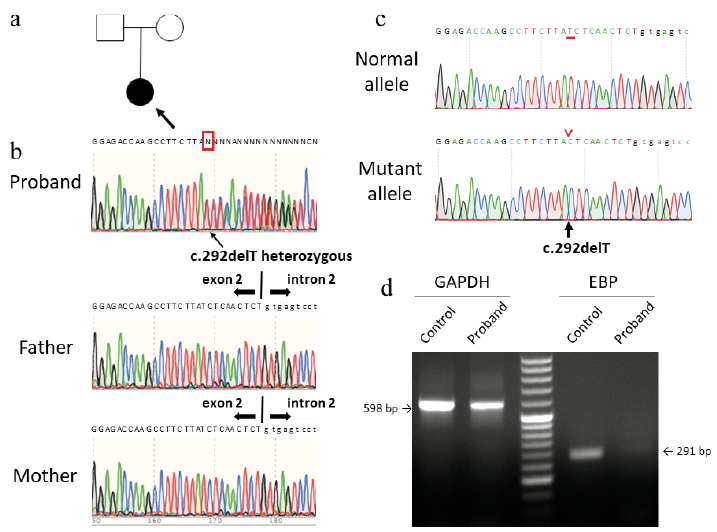 Click to show fullsize
Click to show fullsize