


Department of Dermatology, University Hospital Magdeburg, DE-39120 Magdeburg, Germany. *E-mail: evelyn.gaffal@med.ovgu.de
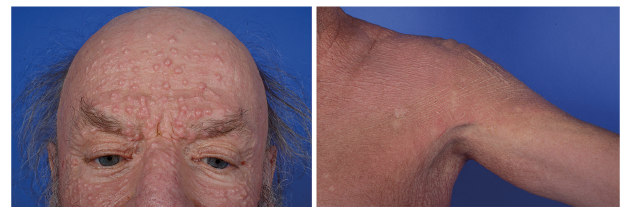
A 63-year-old male patient reported gradual thickening of the skin and development of waxy papules on the dorsal aspects of both hands, with onset of symptoms 8 years previously. In the previous year, the nodules had spread over his entire body (Fig. 1). In addition, he described concurrent development of reduced mobility of his upper extremities, numbness of fingertips and shortness of breath. Trouble swallowing, itch and pain did not occur, family members were reported to be unaffected. Patient history revealed a monoclonal gammopathy of undetermined significance (MGUS).
The current physical examination showed marked sclerosis of the skin, hyperpigmentation and erythematous patches. In addition, linearly distributed prominent papules and nodules of 5-mm diameter were visible on the forehead, shoulders and chest. Histopathological analysis of a skin biopsy from 8 years previously revealed acanthosis, thickened collagen fibres and rich mucin deposits throughout the dermis (Fig. 2, top and mid panel). In comparison, a recent biopsy showed histiocytic-granulomatous infiltrates in the wider dermis (Fig. 2, bottom panel).
Recent blood tests revealed elevated C reactive protein and an accelerated blood sedimentation time, whereas antinuclear antibodies were not detected. Serology for Borrelia antibodies revealed no sign of infection, levels of thyroid hormones were normal.
In a tuning fork test, pallhypaesthesia was observed. Electromyography revealed a decreased amplitude of the tibialis anterior muscle signal. A current thoracic computed tomography (CT) scan revealed a reticular interstitial pattern with thickened septa and ground glass opacity). Furthermore, body plethysmography revealed signs of pulmonary restriction.
What is your diagnosis? Scleromyxoedema? Generalized myxoedema? Systemic sclerosis? Eosinophilic fasciitis?
See next page for answer.
Fig. 1. Current clinical presentation. Clinical images from the face and left shoulder. Patient consent to publish photographs was obtained.
Fig. 2. Histopathological aspects of skin biopsies taken at initial and current presentation. Histopathological sections of a skin punch biopsy taken from the forearm at initial presentation showing acanthosis, thickened collagen fibres and dermal mucin deposits (top and mid panel). In comparison, a recent biopsy showed histiocytic-granulomatous infiltrates in the widened dermis (bottom panel). Top and bottom panels stained with haematoxylin and eosin (H&E), mid-panel stained with Alcian Blue. Scale bar: 250 µm.
Acta Derm Venereol 2021; 101: adv00557.
Diagnosis: Scleromyxoedema
By histopathological correlation, we made the diagnosis of Scleromyxedema. The initially planned therapy with intravenous immunoglobulins could not be performed, due to the patient’s insurance company declining coverage of costs despite multiple appeals. Therefore systemic therapy was commenced with 1 mg per kg body weight prednisolone, administered orally, with tapering over the course of 6 weeks. This therapy resulted in resolution of skin lesions, including induration and suberythroderma. Additional follow-up via CT scans also revealed clearing of the pulmonary sclerosis. However, after discontinuation of therapy, the skin lesions showed rapid relapse within a few weeks. The symptoms showed no improvement on treatment with topical corticosteroids, whereas a short-term therapy with oral prednisolone rapidly achieved disease remission.
A further systemic involvement or progression of MGUS to multiple myeloma could not be detected. The polyneuropathy has shown a stable clinical course to date.
Scleromyxoedema, also referred to as Arndt-Gottron disease, is a rare chronic and progressive primary dermal mucinosis of unknown aetiology. Primarily affected are adults between the ages of 30 and 80 years with an equal distribution between the sexes (1). To date, a hereditary predisposition has not been detected, making a genetic cause unlikely.
Scleromyxoedema, also known as generalized papular and sclerodermoid lichen myxoedematosus, is characterized by the multifocal appearance of symmetrical, waxy papules and nodules in conjunction with sclerosis of the skin by deposition of mucin (2). The cutaneous sclerosis is causative for typical signs of the disease, such as the leonine facies when involving the face and the “Shar-Pei sign” when involving the trunk. Frequently, a brown discoloration and a thinning of body hair is also present.
The typical histopathology of scleromyxoedema is characterized by the triad of: (i) dermal mucin deposits, (ii) fibroblast proliferation, and (iii) thickening of collagen fibres. Approximately one-fifth of all patients show an interstitial granuloma annulare-like pattern (3). This subtype can pose a risk of misdiagnosis if unknown to the pathologist and clinician. The current report shows the first transition between these 2 subtypes.
Cutaneous sclerosis can cause immobilization of adjacent joints in severe cases. In contrast to classical scleroderma, telangiectasias, calcinosis cutis, Raynaud’s syndrome or antinuclear antibodies are rare findings (4). Other symptoms frequently observed in various autoimmune disorders, such as megacapillaries in nailfold capillary microscopy, are also uncommon in scleromyxoedema (5).
The most important differential diagnoses are other mucinoses (generalized myxoedema, pretibial myxoedema, scleroedema, reticular erythematous mucinosis), eosinophilic fasciitis or nephrogenic systemic fibrosis. These can be differentiated via clinical and histological examination as well as blood tests (e. g. blood count, thyroid hormones) (6).
Factors that correlate with the occurrence or clinical course of scleromyxoedema are currently unknown. Interestingly, in almost all cases of scleromyxoedema, an association with paraproteinaemia has been described (1). The transition of MGUS to multiple myeloma or other neoplasia in patients diagnosed with scleromyxoedema is rare, but cannot be excluded (1, 7). Extracutaneous manifestations, including polyneuropathy, thromboembolism, myocardial infarction, and restrictive lung disease, predict the prognosis (1).
Without admission of therapy, scleromyxoedema has a progressive clinical course with a lethality of almost 20% in a mean observation time of 3 years (1). Spontaneous remission has been reported only sporadically (8).
There is currently no approved or evidence-based treatment for scleromyxoedema. Therapeutic options primarily consist of off-label use of diverse immunosuppressive or immunomodulatory substances. Under these, the efficacy of intravenous immunoglobulins has been shown most conclusively. Good clinical regression can also be achieved with systemic therapies using corticosteroids or thalidomide (9). The current case also highlights the efficiency of systemic corticosteroids for pulmonary fibrosis as an extracutaneous manifestation of scleromyxoedema. Regular clinical check-ups are therefore essential to account for the variable course and the risk of therapy-induced adverse reactions, both of which can have lethal outcome (10).


 Click to show fullsize
Click to show fullsize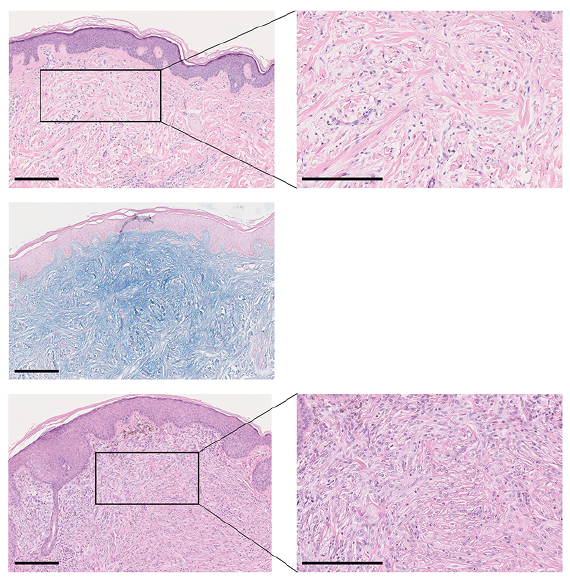 Click to show fullsize
Click to show fullsize