OBJECTIVE: This report describes the design of a study aiming to provide evidence for the extended use of botulinum toxin A (BOTOX®, Allergan Inc.) in focal post-stroke upper and lower limb spasticity and to evaluate the impact of incorporating botulinum toxin A treatment into the rehabilitation of patients with spasticity.
DESIGN: International, prospective, randomized, double-blind, placebo-controlled study with an open-label extension.
METHODS: Approximately 300 adults with a stroke occurring ≥ 3 months before screening, presenting with symptoms and signs of an upper motor neuron syndrome and focal spasticity-related functional impairment, were randomized to botulinum toxin A + standard care or placebo + standard care. Study medication was administered at baseline and again at Week 12 if required, with follow-up to 52 weeks. The primary endpoint was the number of patients who achieved their investigator-rated principal active functional goal (as measured by Goal Attainment Scaling), at 10 weeks after the second injection (Weeks 22−34) or at the 24-week visit if no second injection was administered. Secondary endpoints included changes from baseline in level of goal achievement, health-related quality of life and resource utilization.
CONCLUSION: The BOTOX® Economic Spasticity Trial (BEST) will provide information regarding clinical and cost-effectiveness of botulinum toxin + standard care vs standard care alone in patients with upper and/or lower limb post-stroke spasticity typically seen in clinical practice.
Trial registration: ClinicalTrials.gov number NCT00549783.
Key words: botulinum toxin A; cost-effectiveness; goal attainment scaling; post-stroke spasticity; stroke rehabilitation.
J Rehabil Med 2011; 43: 15–22
Correspondence address: Jörgen Borg, Division of Rehabilitation Medicine, Department of Clinical Sciences, Karolinska Institutet, Danderyd Hospital, SE-182 88 Stockholm, Sweden. E-mail: Jorgen.Borg@ki.se
Submitted January 26, 2010; accepted November 25, 2010
Introduction
The incidence of post-stroke spasticity ranges from 17% to 38% (1−5), with 4–9% of these individuals experiencing disabling spasticity, which is most common in the upper limbs (1, 2). Current guidelines recommend that any patient with motor weakness following stroke should be assessed for the presence of spasticity (6), which is most often graded using the modified Ashworth Scale (MAS) (7). However, the close association of spasticity with other impairments of motor control makes it difficult to determine the extent to which spasticity is the primary cause of an individual’s disability and there is no standardized measure to quantify the specific impact of spasticity (6). In addition, improvements in spasticity may not always occur at the same time as improvements in limb function (8).
Goal attainment scaling (GAS) allows individualization of realistic and feasible goals according to patient needs and expectations (regardless of spasticity presentation), which may encompass everyday activities, self-care or other tasks (6). Measurement of change is performed according to the achievement (or not) of goal(s), reflecting alterations in activity and physical function, which may be more clinically meaningful and sensitive than global measures such as the Barthel Index (9, 10). In addition, the focus is shifted from measuring the extent of disability to the achievement of improvements.
Botulinum toxin A (BoNT-A) is the most widely studied pharmacological intervention for the treatment of focal post-stroke spasticity. When compared with placebo for the treatment of post-stroke spasticity of the wrist and finger in a randomized double-blind trial, a single injection of BoNT-A was associated with a significantly greater proportion of patients achieving an improvement in their principal target of treatment and a significant reduction in disability, lasting for at least 12 weeks following treatment (11). Use of BoNT-A for the management of upper and lower limb focal post-stroke spasticity is common in clinical practice and international guidelines regarding its use in adult spasticity have been published by a number of bodies (12−14).
Post-stroke spasticity is associated with increased direct medical costs during the first year after stroke when compared with stroke survivors not experiencing spasticity (15), but the cost-effectiveness of spasticity treatment has not yet been fully established. The Health Technology Appraisal (HTA) bodies across Europe and North America now require economic evidence based on randomized, controlled trial data including quality of life assessment using validated tools and cost-utility analysis, such as cost per quality-adjusted life year (QALY). In view of this unmet need, the BOTOX® Economic Spasticity Trial (BEST) was specifically designed to evaluate the clinical benefit of including botulinum toxin A in the rehabilitation setting, via a range of patient- and investigator-rated instruments and to provide clinical and cost-effectiveness evidence supporting the use of BoNT-A (BOTOX®, Allergan Inc., Irvine, USA) in upper and lower limb focal post-stroke spasticity to each of these HTA and reimbursement bodies. The rationale and design of the study are presented here.
Study methodology
Study objectives
The primary objective of BEST was to evaluate the effectiveness of BoNT-A + standard care vs placebo + standard care for the treatment of adult post-stroke focal spasticity as measured by the number of patients in each arm achieving their principal active functional goal as determined by the investigator using GAS. Several secondary objectives were also defined (Table I).
| Table I. Secondary study objectives |
| Evaluate the effectiveness of BoNT-A + SC vs placebo + SC for the treatment of adult post-stroke focal spasticity as measured by the number of patients in each arm who achieve their functional goal as determined by the patient. |
| Determine the effectiveness of BoNT-A + SC vs placebo + SC for the treatment of adult post-stroke focal spasticity as measured by the level of functional goal achievement as determined by the patient. |
| Time to functional goal achievement as determined by the physician and patient |
| Evaluate and compare improvement in patients’ quality of life when treated with BoNT-A + SC vs placebo + SC. |
| Collect resource use data from actual clinical practice in the treatment of patients in rehabilitation and other specialized centres. |
| Evaluate and compare level of resource use of patients receiving BoNT-A + SC vs placebo + SC. |
| Evaluate and compare the cost of BoNT-A + SC vs placebo + SC. |
| Identify the drivers of resource use and costs. |
| Compare the cost-effectiveness and cost-utility of BoNT-A + SC vs placebo + SC. |
| Document the occurrence of adverse events during the study. |
| BoNT-A: botulinum toxin A; SC: standard care. |
Study design and setting
BEST was a prospective, multicentre, randomized, double-blind, placebo-controlled Phase IIIb study with an open-label extension, conducted in Germany, Sweden, UK, and Canada (Phase IV). It was designed to reflect normal clinical practice, while allowing efficacy, safety and cost-effectiveness assessments. BEST was conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice and the study protocol was approved by an independent ethics committee at each participating site. Most recruitment sites were in rehabilitation facilities attached to, or in close association with, centres where stroke patients were initially managed.
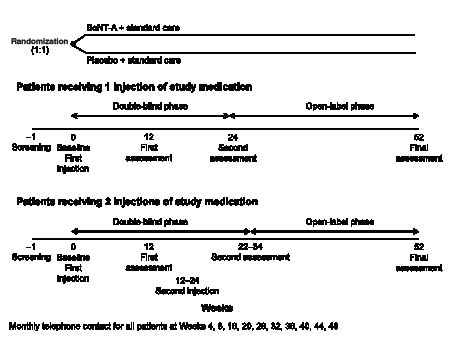
A total of approximately 300 patients were randomized in a 1:1 ratio of BoNT-A + standard care or placebo + standard care, with the treatment arms stratified according to location of spasticity (upper or lower limb) associated with the primary treatment goal set for each patient. The study period included 22–34 weeks of double-blind treatment, at which time the primary endpoint of the study was evaluated (depending on the timing of a possible second injection of BoNT-A), extending to a total of 52 weeks with an open-label phase (during which patients could receive further BoNT-A injections, if required) for resource utilization data collection. A screening visit was followed by the baseline visit (and administration of the first injection of study medication), first assessment (after 12 weeks), optional second injection of study medication (at 12−24 weeks), second assessment (10 weeks after the second injection or 24 weeks if the patient had received only one injection) and follow-up after 52 weeks (Fig. 1).
Study population
Men and women aged 18−85 years were eligible to participate in the study, provided that they had experienced a stroke due to a primary cerebral haemorrhage/infarction or subarachnoid haemorrhage occurring ≥ 3 months before the screening visit and that they were considered by the investigator as suitable for treatment with BoNT-A. In addition, participants were considered by the investigator as likely to experience functional gains following treatment with BoNT-A. This meant for: (i) upper limb spasticity: evidence of preserved antagonist function and ≥ 1 item of the Resistance to Passive Movement Scale (REPAS) (16), with a score of ≥ 1 at the relevant joint(s) for the primary endpoint; or (ii) lower limb spasticity: evidence of preserved standing/ambulation function and ≥ 1 item of REPAS, with a score of ≥ 1 at the relevant joint(s) for the primary endpoint.
Other inclusion criteria for patients entering the study were:
• Not to be of childbearing potential, or agreement to use acceptable and effective birth control during the study.
• To be able to communicate effectively with study personnel (with the help of a third person if required) and willing to follow the study protocol, including attendance at assessment visits and participation in telephone calls with the study investigator.
• At the screening visit, patients were required to give written informed consent to participate in the study.
Patients were excluded from the study if they:
• Had a fixed contracture as a result of spasticity in the limb to be treated in the study or stroke was not the cause of their spasticity.
• Had received previous treatment with botulinum toxin or phenol nerve block in the limbs to be treated during the study.
• Required concomitant intrathecal baclofen or phenol nerve block at any point during the study, or needed systemic aminoglycoside antibiotics or spectinomycin during the period from at least 3 days before until 6 weeks after injection of study medication.
• Had chronic medical conditions that might influence their ability to benefit from treatment or to complete the study, e.g. myasthenia gravis, Eaton Lambert syndrome or conditions affecting the anterior horn cell.
• Had a Mini-Mental State Examination (MMSE) score < 20 (17) or Apathy Evaluation Scale (AES) score > 40 (18).
• Were participating in any other clinical trial or had participated in a clinical trial of a new chemical entity within 6 months prior to the screening visit.
• Had a known hypersensitivity to BoNT-A or excipients of BOTOX®.
• Had an infection at the proposed injection site.
Patients were selected from routine inpatient and outpatient clinical practice and from all levels of the healthcare systems within the participating countries.
Study schedule
The study schedule is shown in Table II. At the baseline visit, the investigator discussed with the patient in which limb they would like to see a functional improvement; this was then designated as the primary assessment limb. The patient and investigator then defined together the principal active functional treatment goal, based on an objective treatment measure (chosen from a list including improvement in the active function of the upper limb to achieve a personal goal, e.g. dressing, writing/typing, feeding, washing, wheelchair propulsion; or improvement in the active function of the lower limb to achieve a personal goal, e.g. ability to sit, assisted transfers, independent transfers, ambulation, climbing stairs) to be achieved by the study intervention for the primary assessment limb. The patient and investigator also defined a secondary active or passive functional treatment goal. The secondary active functional goal was set, either for a different active function in the limb selected for the primary active functional goal, or for an active function in another limb.
The patient was then randomized to treatment with BoNT-A + standard care or placebo + standard care , using a central Interactive Voice Response System (IVRS). Study medication was provided to centres as individual treatment kits, each assigned a unique identification number. On the day of the first treatment injection, individual patients were assigned a treatment kit number provided to the blinded investigators via the IVRS. All investigators, monitors and statisticians involved with the study were blinded for the duration of the trial. Patients were also asked to provide information regarding healthcare resources utilized, e.g. physical or occupational therapies, during the 12 weeks prior to the baseline visit.
| Table II. Study schedule |
| | Screening | Randomization/baseline (1−28 days following screening) | Monthly telephone contacta | Week 12 assessment | Second injection (optional; at weeks 12−24) | Week 24 or 10 weeks after second injection | Week 52/final visit |
| Demographic characteristics | × | | | | | | |
| MMSE (17) | × | | | | | | |
| AES (18) | × | | | | | | |
| REPAS-26 (16) | × | × | | × | × | × | × |
| Medical history | × | | | | | | |
| Features of stroke, including complications
other than spasticity | × | | | | | | |
| Prior/concomitant medication | × | | | | | | |
| Physical examination | | × | | | | | |
| Vital signs | | × | | | | | |
| Urine pregnancy test | | × | | | | | |
| London Handicap Scale | | × | | | | | |
| Barthel Index | | × | | | | | |
| EQ-5D (19)/Short-Form 12 (20) | | × | | × | × | × | × |
| SIS-16 (21) | | × | | × | × | × | × |
| Hamrin & Wohlin Activity Index (22) | | × | | × | × | × | × |
| Pain Scale | | × | | × | × | × | × |
| Goal setting (physician and patient) | | × | | | | | |
| Resource use (economic measures) | | × | | × | × | × | × |
| Resource use (data collection) | | | × | | | | |
| Study medication administration | | × | | | × | | |
| Changes to concomitant medication | | × | | × | × | × | × |
| Adverse events/complications | | × | × | × | × | × | × |
| aPerformed at weeks 4, 8, 16, 20, 28, 32, 36, 40, 44 and 48. AES: Apathy Evaluation Scale; MMSE: Mini-Mental State Examination; REPAS-26: Resistance to Passive Movement Scale 26; EQ-5D: 5-dimension EuroQoL; SIS: Stroke Impact Scale. |
At each visit, with the exception of the screening visit, patients were asked to complete the 5-dimension EuroQoL (EQ-5D) (19), Short-Form-12 health survey (SF-12 v2) (20), 16-item Stroke Impact Scale (SIS-16 v2) (21) and the pain scale, while the investigator completed the Hamrin and Wohlin Activity Index (HWAI) (22) and REPAS-26 (16). Investigators also assessed goal achievement and use of economic and human resources. Patients were monitored for adverse events (AEs) throughout the study.
Treatment protocol
Patients were randomized to either BoNT-A + standard care or placebo (placebo was vacuum-dried saline powder manufactured using the same process as BOTOX®) + standard care treatment arms. Both BoNT-A and placebo were reconstituted prior to use with 2 ml of unpreserved sterile 0.9% (w/v) sodium chloride solution per vial (to achieve a BoNT-A concentration of 50 units/ml). BoNT-A and placebo were administered intramuscularly using sterile needles of gauge 25 or higher according to the situation of the muscles to be injected.
The minimum total dose for each muscle was pre-specified according to consensus from the lead investigators' clinical practice; however, the dose used in individual patients in the study was determined by the investigator, based on his or her experience and normal practice. The exact dosage and number of injection sites was tailored to the individual patient based on the size, number, and location of muscles involved; the severity of spasticity; and the presence of local muscle weakness. Injection details, including muscles injected, number of sites per muscle injected and dosage of study medication were recorded in the case report form (CRF). All clinicians performing the injections were trained in the assessment and injection of BoNT-A for the treatment of post-stroke spasticity and were permitted to use guided injection techniques, e.g. electromyography, nerve stimulation.
BoNT-A should be used as part of a comprehensive therapy programme (13). Patients enrolled in this study received standard care as part of their therapy programme. Standard care was what was routinely available in the individual study centres and agreed between the investigator and patient. This could include physical therapy, occupational therapy, functional electrical stimulation, nursing, oral medication, casting, splinting and home exercises. The components of standard care for each individual patient were documented in the CRF.
Goal attainment scaling
In this study, GAS was measured using a 6-point scale, where −3 represented function that is worse than at the start of treatment, −2 was no change, −1 represented some improvement but did not meet the expected goal, 0 represented goal achievement and +1 or +2 represented over-achievement or exceeding the defined therapeutic goal. An active functional goal was defined as one that required an improvement in the active function of the primary assessment limb to achieve a personal goal, either for the upper limb, e.g. dressing, writing, feeding; or for the lower limb, e.g. walking, climbing stairs. A passive functional goal included relief of symptoms, e.g. pain, spasms; limb posture/positioning; reduction in nurse/carer time required for dressing, hygiene, activities of daily living or other nursing care; or facilitation of services.
To attempt to maximize consistency of the GAS methodology across all study centres and investigators, although all goals were individualized, all investigators were required to complete a course of training with respect to goal identification, setting and scaling prior to participation in the study. In addition, for the first 3 patients enrolled into the study by each investigator, a third party reviewed the goals set to ensure consistency and validity.
Other measures
The other patient-reported outcome measures used in BEST assessed quality of life, changes in function and changes in activity (Table II). The EQ-5D and SF-12 are well-established and validated generic health-related quality of life tools, which have been used in a number of clinical studies involving patients experiencing strokes (19, 20, 23−26). The REPAS-26 is a validated rating scale for assessing resistance to passive movement for a total of 13 passive arm and leg motions on either side of the body (16), while the SIS-16 v2 (21) measures the aspects of stroke recovery that have been identified as important to patients and caregivers as well as stroke experts. The HWAI consists of 16 variables divided into 3 main parts: mental capacity, motor activity, and activities for daily living (ADL) function, with a maximum score of 92 (22).
Patients were monitored for signs and symptoms of AEs throughout the study. Serious AEs (SAEs) were defined as those causing death or those that were life-threatening, required hospitalization or resulted in persistent or significant morbidity. These were reported using a specific SAE form and investigators notified the study monitor of these within 24 hours after they became aware of such events.
Resource utilization and economic evaluation
Resource utilization, as determined by the investigator, was defined in this study as consumption of healthcare and social services resources associated primarily with the treatment of spasticity or related complications, including visits to the study centre, medical/physical interventions, new treatments, diagnostic procedures, concomitant medications prescribed, inpatient stays and complications requiring medical treatment/attention. The study centre contacted the patient every 4 weeks between study visits to obtain details of any contact they had with healthcare professionals and social services since their last visit or telephone contact (described in Table III). In addition, further healthcare resource utilization data was collected in the CRF, e.g. inpatient stays, visits to the study centre, physical interventions, diagnostic procedures, new treatments, concomitant medications and complications.
| Table III. Resource utilization |
| Any change of setting, e.g. home, hospital. |
| Nursing, physical therapy and occupational therapy input. |
| Intensity of caregiver support. |
| Outpatient specialist consultations. |
| Visits to or from general practitioners and other primary healthcare professionals. |
| Changes in non-medical spasticity treatment and concomitant medications. |
| Any equipment prescribed during the initial 24-week study period. |
| Time taken by the patient’s caregiver to accompany them to therapy visits. |
| Distance from medical centres and type of transportation used. |
| Any events affecting the patient’s health, or worsening of any condition present at baseline. |
Costs of resources used were calculated using established methods (27) and 2 perspectives were considered in each country: third-party payer (TPP) and societal. From the TPP perspective, only the portion of costs incurred by the public healthcare system was taken into account. From the societal perspective, costs borne by the healthcare system, transport, caregiver’s time and employers (through sick leave) were included. Unit costs were assigned to the each resource to obtain a cost. Unit costs were derived from a variety of standard sources in the relevant country. Patients’ and caregivers’ lost productivity were calculated based on the cost of labour.
Study endpoints
Primary endpoint. The primary endpoint of the study was the number of patients in each of the two treatment arms who achieved their principal active functional goal (i.e. a score of 0, +1 and +2 inclusive according to GAS), as determined by the investigator, at 10 weeks after the second injection (weeks 22−34) or at the 24-week visit or at time of study withdrawal if no second injection was administered.
Secondary endpoints. A number of secondary endpoints involving secondary active or passive functional goals as measured by GAS, patient-reported outcomes scales and other investigator-reported outcomes were also evaluated during the study (Table IV).
| Table IV. Secondary endpoints |
| Goal attainment scaling |
| The median level of principal functional goal attained in the two treatment arms, as determined by the investigator, at 12 weeks after the first injection, 10 weeks after the second injection, 24 weeks if no second injection has been conducted and at 52 weeks. |
| The number of patients in each of the two treatment arms who achieve their principal functional goal, as determined by the investigator, at 12 weeks after the first injection and at 52 weeks. |
| Time to principal functional goal attainment after the first and second injection, as determined by the investigator. |
| The median level of principal functional goal attained in the two treatment arms, as determined by the patient, at 12 weeks after the first injection, 10 weeks after the second injection, 24 weeks if no second injection has been conducted and at 52 weeks. |
| The number of patients in each of the two treatment arms who achieve their principal functional goal, as determined by the patient, at 12 weeks after the first injection and at 52 weeks. |
| Time to principal functional goal attainment after the first and second injection, as determined by the patient. |
| The number of patients in each of the two treatment arms who achieve their secondary functional goal, as determined by the patient and investigator, at 12 weeks after the first injection, 10 weeks after the second injection, 24 weeks if no second injection has been conducted and at 52 weeks. |
| The median level of secondary functional goal attained in the two treatment arms, as determined by the patient and investigator, at 12 weeks after the first injection,10 weeks after the second injection, 24 weeks if no second injection has been conducted and at 52 weeks. |
| Time to secondary functional goal attainment after the first and second injection, as determined by the patient and investigator. |
| Patient-reported outcomes |
| Change from baseline in EQ-5D and SF-12 scores at 10 weeks after the second injection (24 weeks if no second injection has been conducted) for BoNT-A + SC vs placebo + SC. |
| Change from baseline in EQ-5D and SF-12 scores at 12 and 52 weeks after the first injection for BoNT-A + SC vs placebo + SC. |
| Change from baseline in EQ-5D VAS score at 10 weeks after the second injection (24 weeks if no second injection has been conducted) for BoNT-A + SC vs placebo + SC. |
| Change from baseline in EQ-5D VAS score at 12 and 52 weeks after the first injection for BoNT-A + SC vs placebo + SC. |
| Change from baseline in SF-12 MCS and PCS scores at 10 weeks after the second injection (24 weeks if no second injection has been conducted) for BoNT-A + SC vs placebo + SC. |
| Change from baseline in SF-12 MCS and PCS scores at 12 and 52 weeks after the first injection for BoNT-A + SC vs placebo + SC. |
| Change from baseline in SIS-16 total score at 10 weeks after the second injection (24 weeks if no second injection has been conducted) for BoNT-A + SC vs placebo + SC. |
| Change from baseline in SIS-16 total score at 12 and 52 weeks for BoNT-A + SC vs placebo + SC. |
| Pain Scale score at baseline and subsequent study visits. |
| Investigator-reported clinical outcomes |
| Change from baseline in the mental capacity, motor activity and ADL scores of the Hamrin & Wohlin Activity Index at 10 weeks after the second injection (24 weeks if no second injection has been conducted) for BoNT-A + SC vs placebo + SC. |
| Change from baseline in the mental capacity, motor activity and ADL scores of the Hamrin & Wohlin Activity Index at 12 and 52 weeks for BoNT-A + SC vs placebo + SC. |
| REPAS-26 score at baseline and subsequent study visits. |
| ADL: activities of daily living; BoNT-A : botulinum toxin A; SC: standard care; MCS: Mental Component Summary; PCS: Physical Component Summary; VAS: visual analogue scale; EQ-5D; 5-dimension EuroQoL: SF-12: Short-Form-12 health survey; SIS: Stroke Impact Scale. |
Statistical design and analysis
Sample size calculations
Sample size calculations were performed using nQuery Advisor version 4.0 (Statistical Solutions, Inc., Boston, USA). The sample size calculation for the primary endpoint was based upon an estimate that 80% of patients randomized to BoNT-A + standard care would achieve their principal active functional goal as determined by the investigator, at 10 weeks after the second injection (weeks 22−34) or at the 24-week visit if no second injection was administered vs 50% of patients randomized to placebo + standard care. Setting alpha at 0.05, with a power of 80% to detect a statistically significant difference between treatment arms and assuming a dropout rate of 15%, the maximum number of patients to be enrolled was 210.
The sample size calculation for the secondary endpoint (secondary active functional goal) was similarly based upon 80% of patients randomized to BoNT-A + standard care and 40−50% of patients randomized to placebo + standard care achieving the primary endpoint. It was assumed that the median level of principal functional goal attained would be 0 to 1 for BoNT-A + standard care and −2 to −0.5 for placebo + standard care and a probability of 0.6 that an observation in the placebo + standard care arm would be less than an observation in the BONT-A + standard care arm. With alpha at 0.05, a power of 80% to detect a statistically significant difference between treatment arms and assuming a dropout rate of 15%, the maximum number of patients to be enrolled was 300.
For 300 patients, assuming a true treatment difference in EQ-5D score of 0.1 at 24 weeks, the probability of obtaining an incremental cost-effectiveness ratio (ICER) greater than country-specific thresholds was estimated as 18% for Germany (> €50,000), 7% for Sweden (> €50,000), 7% for the UK (> £30,000) and 22% for Canada (> Can$50,000).
Efficacy variables
The analysis of primary and secondary endpoints was undertaken on an “intention to treat” basis; participants were analyzed in the group to which they were randomized. For the primary endpoint and other endpoints evaluating the number of patients achieving functional goals, logistic regression was employed, allowing for treatment and location of spasticity (upper or lower limb). Summary statistics were calculated for the variables, including the level of functional goal attained and treatment arms were compared using the Wilcoxon two-sample test. Time to goal attainment was calculated as the number of days from the date of the first injection to the date of goal attainment (assessed at 12, 24 and 52 weeks) and treatment groups were compared using Kaplan–Meier survival analysis. Descriptive statistics were calculated for the REPAS-26 scores, SIS-16 v2 scores and the SF-12 v2 MCS and PCS at baseline and throughout the study. Frequencies were reported for each dimension of the EQ-5D, and the effect of treatment on these was investigated using logistic regression. Descriptive statistics were calculated for the health state utilities derived from the EQ-5D and SF-12 v2 and the effect of treatment on these was analyzed using multi-level modelling. QALYs were estimated using utilities derived from the EQ-5D, assuming a linear evolution over the duration of the study.
Utilities
Utilities were collected at all study visits (EQ-5D and SF-12 v2 via the SF-6D [28]): baseline (randomization), week 12, second injection visit (when relevant), week 24 or 10 weeks after the second injection and week 52 (final visit). A linear interpolation was used to calculate QALYs for the base-case analysis. A linear regression model was then used to estimate the effect of treatment on utilities. Independent variables included utility at previous visit, treatment, baseline characteristics, spasticity location, time of assessment, country as fixed effects, a random treatment effect at country level, and random intercept at site levels. Open-label BoNT-A injections were incorporated as an independent covariate in order to predict the utility at 52 weeks for the standard care strategy.
Costs and cost-effectiveness
These calculations were performed in Excel (Microsoft Corporation), with Monte Carlo simulations performed in Crystal Ball version 7.2 (Decisioneering, Denver, USA). The trial observation period of 52 weeks was partitioned into 13 intervals of 4 weeks. For each resource utilization variable, the amount of resource used in each 4-week interval was calculated. Resource utilization over 52 weeks was calculated for each treatment arm, spasticity location and country by summing predicted amounts of resources used in each 4-week interval. A regression model for repeated observations was fitted for each variable representing resource use by interval. Resource utilization over 52 weeks was calculated for each treatment arm, spasticity location and country by summing predicted amounts of resources used in each 4-week interval. Costs were estimated for each patient, summarized by treatment group for each country, and adjusted for age, length of study participation, location, severity, and duration of spasticity, other stroke-associated complications, and other confounders. Descriptive statistics were calculated for resource utilization data used per patient and associated costs over the 52-week study period, and for the major cost drivers an analysis of variance (ANOVA) was applied to test for differences between treatment arms.
The primary cost-effectiveness measure was the incremental cost per additional QALY gained for BoNT-A + standard care vs standard care only (+ placebo). For each country, the study perspective on which the ICER per QALY was calculated from was the TPP perspective, with a 95% confidence interval. The timeframe of the analysis was 52 weeks, with a sensitivity analysis at 24 weeks. Sensitivity analysis was undertaken to assess the impact of key parameters on the results. Other effectiveness outcomes, such as achievement of functional goals, were also be used in subsequent cost-effectiveness analyses.
study Limitations
Although BEST was designed to capture information from usual clinical practice, a number of factors prevented it from completely approximating routine care. These included the number of follow-up visits and telephone calls from the investigators to the patients, which were more frequent than generally occurs in real life and these may have impacted upon treatment outcomes. Standard care from centres of excellence, which were involved in the study, might also have been of a different quality, i.e. with higher minimum requirements, from that delivered in non-specialist settings. Similarly, differences in requirements for standard care may have differed between investigational sites within and between the participating countries. Resource utilization data and their associated costs obtained from the study were therefore subjected to a sensitivity analysis to exclude factors such as the greater frequency and intensity of follow-up.
A 6-point GAS scale, instead of the more commonly used 5-point GAS scale, was employed to allow the demonstration of deterioration from baseline. The follow-up of patients with significant spasticity following stroke affecting function has not been carried out to date and it was therefore important to demonstrate whether patients do deteriorate over the time period of the study. However, while the use of a 6-point GAS scale obviously corresponds to the aims and primary outcome measure of this study, it could limit comparison of the results with those from studies employing other scales. Additionally, data obtained by the BEST modification of the GAS scale cannot be analysed according to T scores. Although the original, symmetrical 5-point GAS scale might be manipulated to include a level of deterioration, the validity of data obtained in that way is still not fully clear (29). From the investigator perspective, learning how to implement GAS required experience of a number of cases prior to being able to set goals with confidence. However, in BEST the investigators were trained and the initial goals set were validated by the study Medical Monitor.
Objective evaluation of goal attainment was possible only at study visits, i.e. 12, 24 and 52 weeks, and this did not allow an accurate record to be made of goal achievement, as the telephone contacts every 4 weeks provided only patients’ subjective views. Instead, the proportion of patients achieving their goals at these time-points was documented, which in itself adds to the somewhat sparse literature about functional improvement in post-stroke spasticity patients.
In conclusion, it is anticipated that BEST will provide a wealth of information regarding the clinical outcomes for post-stroke focal spasticity management with BoNT-A + standard care vs standard care alone, in terms of GAS and other patient- and physician-reported outcomes. In addition, as BEST was designed to be in line with usual clinical practice, it will expand the evidence for the cost-effectiveness of including BoNT-A therapy in the rehabilitation of patients with upper and/or lower limb post-stroke spasticity.
Acknowledgements
Funding: This study is funded by Allergan Ltd. Allergan employees were involved in: (i) the design and conduct of the study; and (ii) the collection, management, analysis, and interpretation of the data. The authors had sole control over the preparation, review, and approval of this manuscript. Editorial assistance for preparation of this manuscript was provided by Michelle Preston and Right Angle Communications.
Conflicts of interest: Professor Jörgen Borg has been the recipient of honoraria and fees for presentations at meetings and congresses and for participating in Advisory Boards.
Professor Anthony B. Ward has participated in research studies, for which unrestricted grants have been provided by Allergan. He has been the recipient of honoraria and fees for presentations at meetings and congresses and for participating in Advisory Boards. He has also received, in the past, honoraria and fees from Ipsen, Medtronic and Merz for presentations at meetings and congresses.
Professor Jörg Wissel has participated in research studies, for which unrestricted grants have been provided by Allergan, Elan, Merz and Ipsen. He has been the recipient of honoraria and fees for presentations at meetings and congresses and for participating in Advisory Boards from Allergan, Eisai, Ipsen, Merz and Medtronic.
Professor Jai Kulkarni have been the recipients of honoraria and fees from Allergan and Ipsen for presentations at meetings and congresses.
Dr Mohamed Sakel have been the recipients of honoraria and fees from Allergan and Ipsen for presentations at meetings and congresses.
Dr Per Ertzgaard has been the recipient of honoraria and fees for presentations at meetings and congresses and for participating in Advisory Boards. He has also received honoraria and fees from Medtronic for presentations at meetings and congresses.
Dr Per Åkerlund has no conflicts of commercial interest in this study.
Dr Iris Reuter has received honoraria and fees for presentations at meetings and congresses and for participating in Advisory Boards. She has also received, in the past, honoraria and fess from Ipsen, Merz and Medtronic for presentations at meetings and congresses.
Dr Christoph Herrmann has participated in research studies, for which unrestricted grants have been provided by Allergan. He has received, in the past, honoraria and fees from Allergan, Ipsen, Medtronic and Merz for presentations at meetings and congresses.
Dr Lalith Satkunam has participated in educational and clinical activities for which unrestricted grants have been provided by Allergan and Medtronic. He has received expenses only for participation in Advisory Boards and CME activities from Allergan and Merz.
Dr Theodore Wein has participated in research studies for which unrestricted grants have been provided by: Allergan, Sanofi, Bristol Myers Squibb, Pfizer and the National Institutes of Health. He has received honoraria for participating in congresses, Advisory Boards and accredited CME activities for: Allergan, Bristol Myers Squibb, Sanofi, Pfizer and Servier.
BEST Study Group: Dr Per Åkerlund, Falu Lasarett, Falun, Sweden; Dr Tun Aung, West Park Rehabilitation Hospital, Wolverhampton, UK; Dr Janne Biörklund, Rehabcentrum Lund-Orup, Lund, Sweden; Professor Jörgen Borg, Karolinska Institutet, Danderyd Hospital, Stockholm, Sweden; Professor Dr Andres Ceballos-Baumann, Neurologisches Krankenhaus München, München, Germany; Dr Christian Dohle, MEDIAN Klinik Berlin, Abteilung für Neurologische Rehabilitation, Charité – Universitätsmedizin, Berlin, Germany; Dr Micael Edblom, Länssjukhuset Ryhov, Jönköping, Sweden; Dr Per Ertzgaard, Linköping University Hospital, Linköping, Sweden; Dr Naseer Haboubi, Linden Lodge Rehabilitation Centre, Nottingham, UK; Dr Anders Häggström, Rehabiliteringsmedicinska Kliniken, Örebro, Sweden; Dr Christoph Herrmann, Asklepios Kliniken Schildautal Kliniken für Neurologische Rehabilitation und Frührehabilitation, Seesen, Germany; Professor Stefan Hesse, Medical Park Berlin Humboldtmühle Neurologie, Berlin, Germany; Dr Jai Kulkarni, Manchester Royal Infirmary, Manchester, UK; Dr Kjell Kullander, Karolinska Universitetssjukhuset, Huddinge, Sweden; Dr Lal Landham, Medway Maritime Hospital, Gillingham, UK; Dr Kristina Lindgren, Centralsjukhuset, Neuro-och rehabkliniken, Karlstad, Sweden; Dr Sabine Mehnert, Justus Liebig Universität, Giessen, Germany; Dr Friedmann Müller, Neurologische Klinik Bad Aibling, Bad Aibling, Germany; Dr Margaret Phillips, Derby City Hospital, Derby, UK; PD Dr Thomas Platz, Neurologisches Rehabilitationszentrum Greifswald, Greifswald, Germany; Dr Robert Prempeh, Falkirk & District Royal Infirmary, Falkirk, UK; Dr Iris Reuter, Justus Liebig Universität, Giessen, Germany; Dr Chris Roy, Southern General Hospital, Glasgow, UK; Dr Mohamed Sakel, Kent & Canterbury Hospital, Canterbury, UK; Dr Patrik Säterö, SU/Högsbo sjukhus, Göteberg, Sweden; Dr Lalith Satkunam, Glenrose Rehabilitation Hospital, Edmonton, Canada; Dr Wilfried Schupp, Fachklinik Herzogenaurach, Herzogenaurach, Germany; Dr Klaus Seigel, Akademiska sjukhuset, Uppsala, Sweden; Dr Satyendra Sharma, Sunnybrook Health Science Centre, Toronto, Canada; Dr Christian van der Ven, Neurologisches Rehabilitationszentrum Bonn Godeshöhe, Bonn, Germany; Dr Martina Walsh, West Midlands Rehabilitation Centre, Birmingham, UK; Professor Anthony Ward, North Staffordshire Rehabilitation Centre, Stoke-on-Trent, UK; Dr Theodore Wein, Montreal General Hospital, Montreal, Canada; Professor Dr Jörg Wissel, Neurologische Rehabilitationsklinik, Beelitz-Heilstätten, Germany.
References