Carl-Henrik Nordström, MD, PhD, Troels Halfeld Nielsen, MD and Anne Jacobsen, MD
Department of Neurosurgery, Odense University Hospital, Odense, Denmark
OBJECTIVE: To describe innovations in neurocritical care originating from university hospitals in southern Scandinavia over a period of 50 years.
Discussion: Several techniques and strategies that are now included in clinical routine were initially developed in southern Scandinavia: continuous recording of intracranial pressure, monitoring of cerebral blood flow, analyses of cerebral energy metabolism under physiological and pathological conditions, and intracerebral microdialysis with bedside biochemical analysis and display of data. This background and, in particular, knowledge of the physiological prerequisites for water transport across the blood–brain barrier and the regulation of brain volume constituted the basis for the “Lund Concept” for treatment of increased intracranial pressure. The development of neurocritical care has resulted in a dramatic decrease in mortality for patients with severe traumatic brain injury.
CONCLUSION: The focus in the future may be on improved biochemical supervision at the bedside to avoid secondary episodes of ischaemia and to identify and treat secondary non-ischaemic mitochondrial dysfunction. As mortality has decreased, demand for qualified post-traumatic rehabilitation has increased. Further improvements will necessitate close cooperation between critical care physicians, neurosurgeons and specialists in rehabilitation medicine.
Key words: Critical care; intracranial pressure; craniocerebral trauma; cerebrovascular circulation; microdialysis.
J Rehabil Med 2013; 45: 00–00
Correspondence address: Carl-Henrik Nordström, Department of Neurosurgery, Odense University Hospital, Denmark. E-mail: carl-henrik.nordstrom@med.lu.se
Accepted February 19, 2013
INTRODUCTION
Critical, or intensive, care medicine is defined as a branch of medicine concerned with the diagnosis and treatment of life-threatening conditions requiring invasive monitoring and sophisticated pharmacological or instrumental organ support. To accomplish this task it is necessary to have specially educated personnel as well as specially designed intensive care units. The first critical care unit in the world opened in Copenhagen in December 1953 (1).
The pivotal point for the development of intensive care occurred during the 1952 polio epidemic in Copenhagen. When 27 out of 31 poliomyelitis patients with respiratory involvement had died during the first weeks of the epidemic the anaesthetist Bjørn Ibsen (1915–2007) was asked for advice. He quickly realized that the patients died from respiratory insufficiency with carbon dioxide retention. On the 27 August 1952 he initiated protracted positive pressure ventilation with tracheal intubation via a tracheostomy in the first patient. For several weeks he had 40–70 patients requiring continuous or intermittent bag ventilation. To accomplish this, approximately 200 medical students were enlisted to ventilate the patients manually. It was reported that this management decreased mortality from close to 90% to approximately 25% (1–3). As a result of this remarkable achievement the first intensive care unit was opened by Dr Ibsen in the Observation Room at Kommunehospitalet (the Municipal Hospital) in Copenhagen in 1953. However, it was almost 4 decades before the first intensive care unit dedicated to neurocritical care was opened. This paper reviews the many techniques and strategies now used worldwide within neurocritical care that originate from southern Scandinavia.
INTRACRANIAL PRESSURE MONITORING
Neurocritical care is characterized by monitoring techniques necessary for identifying secondary cerebral adverse events and for the evaluation of specific therapies. Among these techniques intracranial pressure (ICP) monitoring is by far the most important. The technique for continuous monitoring of ICP was developed during the 1950s at the Department of Neurosurgery, Lund University Hospital by Nils Lundberg (1908–2002) (Fig. 1). His experiences were collected in a doctoral thesis (4) that was defended on 11 February 1961. Both faculty opponents considered the thesis a very valuable scientific contribution, but opposition ex auditorio claimed that the work was unethical and that it had led to mortality in some patients. Due to this criticism the evaluation board had extensive discussions for 3 days [sic] before the thesis was accepted with very high marks (5).
Fig. 1. Nils Lundberg (1908–2002), Professor of Neurosurgery at Lund University Hospital 1962–1974, introduced the technique for continuous monitoring of intracranial pressure.

In 1959 Lundberg had presented data showing that controlled hyperventilation led to a reduction in increased ICP (6) and in his doctoral thesis he documented that intravenous (i.v.) infusion of hypertonic solutions (e.g. urea, mannitol) could also be used for this purpose (4). Lundberg also described the variations in ICP that occurred under physiological and pathophysiological conditions. Most importantly, he described the A-waves (“plateau waves”) that are observed primarily in patients with intracranial mass lesions and may precede the final brainstem incarceration (4). The first study of ICP in patients with severe brain trauma was published in 1965 by Lundberg and collaborators (7). The fact that the thesis and subsequent studies had proven that protracted continuous monitoring of ICP was possible without serious complications (8) meant that his technique became the cornerstone for the development of neurocritical care.
REGIONAL CEREBRAL BLOOD FLOW
In the 1940s Kety & Schmidt (9), in a series of publications, had demonstrated that it was possible to measure human global cerebral blood flow (CBF) by the use of the inert gas nitrous oxide. The Danish physiologist Niels Lassen (1926–1997) extended the measurement of global CBF by including radioactive isotopes (10). In collaboration with the Swedish neurophysiologist David Ingvar (1924–2000) he presented a novel method for the measurement of regional cerebral blood flow (rCBF) based on intracarotid injection of the radioactive substances 85Krypton or 133Xenon dissolved in saline. Clearance of the isotopes was recorded by extracranial detectors (11, 12). The combined studies, performed in Copenhagen and Lund, introduced a new era in experimental and clinical brain research. The Lassen group in Copenhagen focused their interest on the physiology and pathophysiology of the cerebral circulation, while Ingvar and his collaborators in Lund studied predominantly neuropsychological and neuropsychiatric problems.
In the original technique for measuring rCBF the radioactive isotope was infused via the carotid artery. During the early 1970s this technique was also introduced in studies of patients with severe brain trauma (13, 14). Among these pioneers were Jørn Overgaard (1927–1984) and collaborators at the University Hospital in Odense (15) and Georg Cold and collaborators at the University Hospital in Aarhus (16–18). Many important observations were published in their studies, but 3 of these primarily influenced neurocritical care. First, global CBF was found to be unrelated to neurological state and clinical outcome (14, 15). Secondly, impaired cerebral pressure autoregulation was frequently observed and was not related to clinical outcome (15, 18). Thirdly, cerebrovascular CO2 reactivity was often impaired during the acute phase after trauma, in particular in patients with very severe lesions and a poor outcome (15, 17). However, the technique of infusing the radioactive tracer into the carotid artery was a severe limitation for repeated measurements and prevented the technique from being introduced in clinical practice within critical care. For further development it was also necessary to combine measurements of CBF and aspects of cerebral energy metabolism.
LABORATORY FOR EXPERIMENTAL BRAIN RESEARCH IN LUND
In the early 1960s Nils Lundberg was contacted by the young, promising neuroscientist Bo Siesjö. The contact led to the creation of the Laboratory for Experimental Brain Research, which came to be of fundamental importance for the development of neurocritical care. The laboratory originally focused on the regulation of extra- and intra-cellular acid–base relations in the brain (19). Techniques for quantitative measurements of global and regional CBF in small animals were gradually developed and, simultaneously, advanced micro-techniques for determination of a large number of biochemical variables were introduced. It was then possible for a whole generation of young clinicians to study complicated problems related to neurocritical care in experimental models: cerebral ischaemia, hypoxia and hypoglycaemia; induced epileptic seizures; hyper- and hypo-thermia; mechanisms of irreversible cell damage leading to cell death. Neurosurgeons, neurologists, surgeons, paediatricians and anaesthetists received their scientific education in the laboratory and presented their doctoral theses here. At that time the Laboratory for Experimental Brain Research was generally considered one of the leading laboratories in the world, and international neuroscientists regularly visited the laboratory. Many aspects of the work and the results obtained were summarized by Dr Siesjö in a series of review articles in clinical scientific papers and in a comprehensive textbook (20–22).
For many years the results of these experimental studies were of importance mainly for the understanding of the pathophysiological processes in neurocritical care. It was not until the technique of microdialysis was introduced in the 1990s as a bedside method of surveillance that cerebral biochemistry was integrated into the treatment of individual patients.
BEDSIDE MEASUREMENT OF CEREBRAL BLOOD FLOW, VASOREACTIVITY AND OXYGEN CONSUMPTION
The anaesthetist Kenneth Messeter (1932–2003) was of crucial importance for the development of new therapeutic strategies in severe head injuries. Dr Messeter had received his scientific education at the Laboratory for Experimental Brain Research, and he introduced a technique for measurement of CBF that could be used at the bedside. The tracer substance 133Xenon dissolved in saline was injected intravenously, followed by a rapid injection of 20 ml isotonic saline. Clearance of the tracer was monitored from both parietotemporal regions by two scintillation detectors and from the expired air. The CBF data were automatically calculated from the clearance curves by conventional bicompartmental analysis and a delayed-start fit time (23).
Based on previous clinical experiences from Odense and Aarhus, and experimental studies at the Laboratory for Experimental Brain Research it was decided to focus on 3 areas of interest: (i) cerebral vasoreactivity, as determined from changes in CBF due to a change in PaCO2 induced by hyperventilation; (ii) CBF after i.v. injection of phenobarbital to achieve a burst-suppression pattern on EEG; (iii) measurement of cerebral oxygen consumption (CMRO2) after induced barbiturate coma. The studies showed that, in patients with preserved cerebral vasoreactivity to hyperventilation (CO2-reactivity), barbiturate coma therapy was accompanied by a decrease in ICP due to a reduction in CBF with a parallel decrease in CMRO2 (24). In patients with impaired CO2-reactivity, these changes in CBF and CMRO2 were not obtained (24). As a lasting decrease in ICP was not obtained the prognosis was extremely bad in the latter group.
Introduction of the barbiturate coma therapy was paralleled by efforts to evaluate the efficacy of improvements in critical care. Inspired by Dr Messeter a careful follow-up study of all patients treated for severe traumatic brain lesions had previously been performed at the Department of Neurosurgery in Lund. The patients were selected according to the Glasgow definition of severe brain trauma (25) and clinical outcome was judged according to the Glasgow Outcome Scale (GOS) (26). The results are shown in Table I. The results in Lund were, at that time, equal to the experiences in other large international studies (27, 28). After the introduction of a standardized protocol for critical care in these patients (including induction of barbiturate coma in selected patients), the outcome results improved significantly (Table I) (29). In particular, it was noted that the reduction in mortality did not lead to an increase in the proportion of patients remaining in severe disability or a vegetative state.
|
Table I. Data from the Department of Neurosurgery, University Hospital in Lund during 3 time periods (27, 29, 37) |
|||
|
Outcome |
1977–1982 n = 425 Original conventional therapy |
1983–1984 n = 162 Barbiturate coma therapy |
1989–1994 n = 53 Lund Concept |
|
Good recovery/ moderate disability, % |
39 |
54 |
79 |
|
Severe disability/ vegetative state, % |
13 |
11 |
13 |
|
Dead, % |
48 |
35 |
8 |
The department now focused on two problems. First, the fact that a large proportion of our patients were classified as good recovery/moderate disability did not imply that they could return to a normal life (30, 31). This experience indicated that, in the future, more interest should be directed to the rehabilitation period. Secondly, as a subgroup of patients with extremely high mortality could be defined from objective, physiological data (CO2-reactivity) it was considered ethically motivated to introduce a completely new therapy in these patients. This subgroup consisted of patients with high ICP (> 20 mmHg) with impaired cerebrovascular reactivity to changes in PaCO2 (32).
LUND CONCEPT
As a subgroup of patients with extremely high mortality had been identified (32) the interest was directed towards the basic principles for reduction in increased ICP. Here the physiologist and anaesthetist Per-Olof Grände contributed with knowledge regarding physiological principles for tissue volume regulation under experimental conditions (33). These principles, which constituted the scientific basis for the “Lund Concept” (34), were to a large extent based on extensive basic studies previously presented by the US physiologist JD Fenstermacher (35).
Volume regulation of the brain is, as in other organs, determined mainly by mechanisms controlling the water exchange across the capillaries. However, the brain differs from all other organs in its highly sophisticated capillary membrane function, the blood–brain barrier (BBB). In addition to its other physiological functions the BBB is the most important regulator of cerebral volume (35). The principles are illustrated schematically in Fig. 2 for 3 hypothetical situations: (i) the normal brain with intact BBB; (ii) the injured brain with BBB permeable for crystalloids but not colloids; (iii) the injured brain with a ruptured BBB permeable for crystalloids as well as colloids.
Due to the BBB the prerequisite for water transport across cerebral capillaries is different from all other tissues. The intact BBB is impermeable for the two major solutes of biological fluids (Na+ and Cl–) (Fig. 2A). Water passing the BBB in any direction will thus be virtually devoid of crystalloids and an opposing osmotic gradient, which counteracts further fluid movement, will immediately be created. In all other tissues transcapillary water transport is governed by the balance between intracapillary hydrostatic pressure and blood colloidal pressure, both amounting to approximately 20 mmHg. As the total crystalloid osmotic pressure is approximately 5700 mmHg intracapillary pressure as well as variations in blood colloidal osmotic pressure is of very limited importance provided the BBB is intact. Under physiological conditions the brain is also protected from increases in intracapillary hydrostatic pressure during an increase in mean arterial blood pressure (MAP) and cerebral perfusion pressure (CPP) by autoregulation of CBF. Accordingly, the brain is under normal conditions effectively protected from variations in volume, which is extremely important as it is surrounded by the rigid skull.
In the injured brain the BBB may be partly ruptured and permeable to crystalloids. In these patients cerebral autoregulation is also often impaired (15, 18) and cerebral transcapillary water flux will then, as in all other tissues, behave according to the Starling equilibrium: water transport is determined by the balance between the differences in hydrostatic and colloidal osmotic pressure (Fig. 2B). If these patients develop increased ICP, the adequate treatment is to reabsorb water over the capillary endothelium by decreasing intracapillary hydrostatic pressure, e.g. by a pharmacological reduction in MAP, while colloidal osmotic pressure should be kept within physiological limits.
If the BBB of the injured brain is more or less completely ruptured, then large colloidal molecules will also pass into the interstitium. In this situation transcapillary water transport is determined by the difference in hydrostatic pressure across the capillary wall (Fig. 2C). This situation is not infrequently experienced by neurosurgeons: when the skull bone and the dura are opened in a patient with a serious cerebral insult the momentary increase in the transcapillary pressure gradient may cause a rapid increase in transcapillary water transport, pronounced brain swelling and a bulging of the brain outside the craniotomy.
The physiological and pathophysiological considerations schematically depicted in Fig. 2 constituted the background for the “Lund Concept” for treatment of elevated ICP (36). Introduction of the “Lund Concept” resulted in a dramatic decrease in mortality at the Department of Neurosurgery in Lund (Table I). The marked decrease in mortality was associated with significant increases in the groups of “good recovery” and “moderate disability”, but did not increase the number of patients in the groups of “severe disability” or “vegetative state”. In the original study, which included a selected group of patients with very severe traumatic brain lesion and ICP above 25 mmHg in spite of conventional treatment, the mortality decreased from 47% to 8% (37). Similar good results were published from other Swedish neurosurgical centres adopting the “Lund Concept” (38, 39). These studies were all based on comparisons with outcome in historical control patients. Recently, however, a prospective, randomized study showed a similar improvement in patients treated according to the “Lund Concept” (40).

Fig. 2. Water exchange across cerebral capillaries in 3 hypothetical situations: (A) the normal brain with intact blood–brain barrier (BBB); (B) the injured brain with a BBB permeable for crystalloids but not colloids; (C) the injured brain with a ruptured BBB permeable for crystalloids as well as colloids. Grey area: crystalloids in the capillary; open circles: large (colloidal) molecules; filled circles: blood cells.
The fact that a pronounced reduction in mortality had been achieved without increasing the number of patients in a vegetative state or with remaining severe disability did not, however, decrease the demands for qualified rehabilitation after the acute phase. The results of a careful follow-up study documented that the improvements in neurocritical care increased the demands for rehabilitation in patients classified as good recovery/moderate disability (41). In an attempt to quantify the specific effects of rehabilitation data obtained from neuropsychological tests and occupational performance (assessment of motor and process skills, AMPS) was evaluated on admission to the rehabilitation centre and compared 3, 6 and 12 months later (42). The study showed that AMPS gave a different view of the patient’s restitution than neuropsychological tests and might be a better indicator of the patient’s ability to resume independent living. Furthermore, the study indicated a direct positive effect of rehabilitation on AMPS and the deterioration of process skills post-rehabilitation suggested that lasting contact in an outpatient setting might facilitate return to social life (42).
In spite of the favourable clinical results obtained, an important problem related to the “Lund Concept” remained. As a pharmacologically induced reduction in MAP is a fundamental component of the concept, it was compulsory to define the lower acceptable limit for CPP in the individual patient. If this limit is unknown there is a risk that the therapy might lead to ischaemia, in particular in the sensitive penumbra zone surrounding focal brain lesions. To accomplish this task it was necessary to monitor cerebral energy metabolism at the beside during neurocritical care.
BEDSIDE BIOCHEMICAL MONITORING BY MICRODIALYSIS
Microdialysis was introduced almost 40 years ago by Urban Ungerstedt (43) at the Karolinska Institute in Stockholm primarily for experimental monitoring of the animal brain. Due to our collaboration with Dr Ungerstedt, in 1996 we had the opportunity to pioneer microdialysis with bedside biochemical analysis and display of the data.
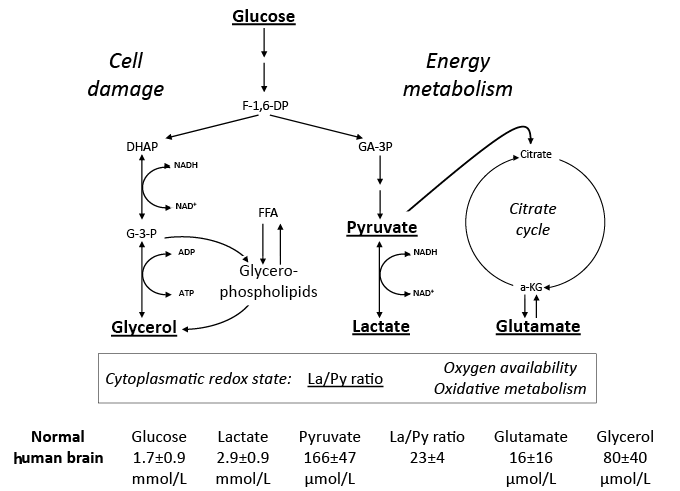
The basic idea of microdialysis is to mimic the function of a blood capillary by inserting a thin dialysis tube (< 0.6 mm) into the tissue. The membranous wall of the tube allows free diffusion of water and solutes between the surrounding interstitial fluid and the perfused solution (perfusate). The concentration gradients between the interstitial fluid and the perfusate constitute the driving force for diffusion. The molecular weight of the molecules being sampled is limited by the pore size of the dialysis membrane (cut-off). The perfusate flows along the dialysis membrane slowly and at a constant speed, and the sample (dialysate) is collected and analysed biochemically. Accordingly, the technique allows analysis of virtually all chemical compounds that pass through the dialysis membrane. During clinical conditions these analyses are performed by utilizing conventional enzymatic techniques (44). The bedside analyses focus on two aspects of cerebral metabolism: variables involved in cerebral energy metabolism and variables indicating threatening energy crises and degradation of cellular membranes. Fig. 3 provides a schematic picture of the chemistry relevant for routine neurocritical care. As discussed before, much of the biochemical changes during various patho-physiological conditions had been clarified in animal experiments at the Laboratory for Experimental Brain Research decades before (20–22). However, it should be recognized that the biochemical information during neurocritical care is obtained exclusively from the extracellular space, while the animal studies were performed on homogenized whole brain.
Under normal circumstances glucose is the sole substrate for cerebral energy metabolism. The interstitial level obtained by microdialysis reflects the relationship between the delivery of glucose from the capillaries and its uptake into the cells. In the cellular cytoplasm it is stepwise degraded to pyruvate (anaerobic glycolysis). Under physiological conditions (i.e. sufficient oxygenation, functioning mitochondria) most of the pyruvate is in the mitochondria degraded completely to CO2 and H2O, where most of the energy released is transferred into ATP. Under normal conditions approximately 5% of the pyruvate is converted to lactate by lactate dehydrogenase in the cytoplasm (Fig. 3). The reaction is a reversible equilibrium reaction reflecting cytoplasmatic redox state, i.e. tissue oxygenation and mitochondrial function. When tissue oxygenation is insufficient, or when the mitochondrial function is impaired the lactate/pyruvate (La/Py) ratio will increase. As lactate and pyruvate are equally permeable across the cell membranes the La/Py ratio measured in the intercellular space will give true information regarding the cytoplasmatic redox state.
Fig. 3. Simplified diagram of cerebral intermediary metabolism, with a focus on the glycolytic chain and its relation to glycerol and glycerophospholipids and to the citric acid cycle (Krebs cycle). F-1,6-DP: fructose-1,6-diposphate; DHAP: dihydroxyacetone-phosphate; GA-3P: glyceraldehyde-3-phosphate; G-3-P: glycerol-3-phosphate; FFA: free fatty acids; α-KG: α-ketoglutarate. Underlined metabolites are measured at the bedside with enzymatic techniques. References levels of the various metabolites for normal human brain obtained from (44).
Secondary cerebral ischaemia was originally considered the main target for clinical microdialysis, and most clinical studies initially focused on this problem in conditions such as severe brain trauma (45), subarachnoid (46) and intracerebral haemorrhage (47). Tissue microdialysis, by necessity, provides biochemical information from a very small zone surrounding the catheter. As cerebral energy metabolism varies in different brain regions the positioning of the catheter is of paramount importance (48). Furthermore, as microdialysis is a very local technique it is usually futile to correlate changes observed in biochemistry with the eventual clinical outcome. Data obtained from microdialysis provide information about local tissue damage and local tissue outcome. Accordingly, to be of clinical relevance it is necessary to localize the position of the microdialysis catheter in relation to the pathological process and, if necessary, insert multiple intracerebral catheters (48). When cerebral microdialysis is used in an optimal way it offers a possibility to detect secondary cerebral ischaemia before it is revealed by global techniques, to start adequate therapy early, and thus prevent further tissue damage (49).
As mentioned above, clinical microdialysis was introduced at the Department of Neurosurgery in Lund in order to monitor patients with severe brain trauma treated according to the “Lund Concept” and to ensure that the decrease in CPP did not cause secondary brain damage. In a comprehensive study it was documented that cerebral energy metabolism usually tolerated a decrease in CPP to our previously defined lower level of 50 mmHg (50). However, this level should not be regarded as fixed: it varies between different patients and the lower acceptable level for the individual patient can only be assessed by monitoring cerebral energy metabolism in the sensitive penumbra zone.
BEDSIDE DIAGNOSIS OF CEREBRAL ISCHAEMIA AND MITOCHONDRIAL DYSFUNCTION
Clinical microdialysis initially focused on identifying, and avoiding, episodes of secondary clinical ischaemia. However, clinical studies from several neurocritical care units indicated that prolonged disturbance of cerebral energy metabolism, including increase in the La/Py ratio, was often not due to ischaemia (51). The condition observed was generally named “hyperglycolysis” or “metabolic crisis”.
By the 1970s animal experiments had shown that transient cerebral ischaemia often lead to a prolonged period of mitochondrial dysfunction (52, 53). Although the pattern of variables related to cerebral energy metabolism was documented in many studies the importance of these observations were not noticed. We have recently completed a series of animal experiments at the Department of Neurosurgery, Odense University Hospital. The experiments were performed to define the biochemical pattern obtained during mitochondrial dysfunction in order to distinguish and separate it from cerebral ischaemia (54, 55). The results are shown schematically in Fig. 4.
Fig. 4 illustrates the biochemical changes and the change in tissue oxygenation (PtiO2) in 3 situations. In cerebral ischaemia the cessation of blood flow and decrease in PtiO2 causes a very rapid increase in La/Py ratio (Fig. 4). As the cerebral delivery of substrate for energy metabolism (glucose) is also interrupted, pyruvate decreases to a very low level. As a result the La/Py ratio increases to extremely high levels. In mitochondrial dysfunction PtiO2 is unchanged but, due to impaired mitochondrial function, oxidative metabolism is insufficient to meet the energy demands. The increase in glycolytic rate causes a massive production of lactate and increase in the La/Py ratio although tissue pyruvate remains at a normal level or increases slightly (Fig. 4). For comparison, Fig. 4 also illustrates the situation during arousal/awakening. In this situation the increase in energy consumption is met by an increase in oxidative metabolism, lactate and pyruvate increase in parallel, and the La/Py ratio remains constant.
Under clinical conditions an increase in La/Py ratio may be caused by a variety of mechanisms (56). Irrespective of the mechanisms underlying mitochondrial dysfunction, a beneficial therapeutic intervention would probably be reflected in normalization of the biochemical variables analysed and displayed at the beside. There is reason to believe that drugs that are effective in mitochondrial dysfunction will soon be clinically available. One example is cyclosporine A, which is thought to decrease mitochondrial damage by blocking opening of the mitochondrial permeability transition pore (57). The protective effect of cyclosporine in cerebral ischaemia was first described at the Laboratory for Experimental Brain Research (58), and the drug is presently being prepared for the first clinical trial in patients with traumatic brain injury.
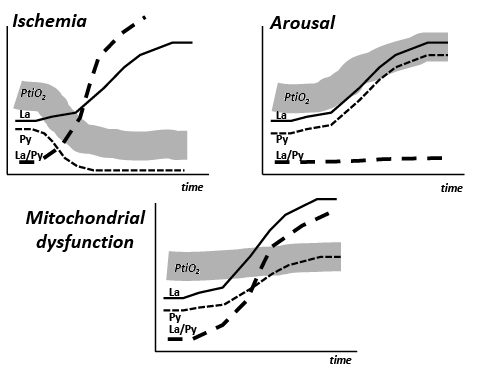
Fig. 4. Cerebral tissue oxygenation (PtiO2) and changes in the levels of lactate (La), pyruvate (Py), and the lactate/pyruvate ratio (La/Py) in 3 conditions: ischaemia, arousal, and mitochondrial dysfunction.
The development of new pharmacological therapies may improve outcome after severe brain trauma. Since mortality has already been reduced to a very low level with conventional management (Table I), it is unlikely that new drugs will reduce it further. The hope is rather that new therapies may prevent secondary damage in the sensitive penumbra zone, thereby improving quality of life for survivors. The possible positive effect will be difficult to evaluate objectively and will necessitate close cooperation between critical care physicians, neurosurgeons and specialists in rehabilitation medicine. For evaluation of the efficacy of new therapies it will also be necessary to improve techniques for physiological and biochemical evaluation of tissue outcome.
CONCLUSION
This review has focused on important innovations within neurocritical care that originate from a few university hospitals in southern Scandinavia: the first unit for intensive care, the development of techniques for measuring ICP, CBF and analyses of cerebral energy metabolism and the introduction of microdialysis as a routine clinical technique leading to the possibility to diagnose and separate ischaemia and mitochondrial dysfunction at the bedside.
In this review we have demonstrated that the introduction of new physiological and biochemical monitoring techniques have increased our knowledge of the complex pathophysiological situation in severe brain trauma. The knowledge has resulted in a new therapeutic principle, the “Lund Concept” for treatment of brain oedema, and a significant improvement in clinical outcome. Furthermore, we have noted that a decrease in mortality may result in an increasing number of patients in need of qualified rehabilitation. In our experience, the examination and evaluation of the patients by the rehabilitation team already during the latter phase of neurocritical care facilitates cooperation between the departments and a smooth transfer. Our data also indicate that following the initial treatment within the rehabilitation centre a continuous, protracted and structured contact with the outpatient’s department will improve the long-term clinical result.
Improvements within critical care were linked to the introduction of new techniques to evaluate the physiological and biochemical state of the patients. New monitoring techniques are, however, necessary not only for the development and evaluation of new treatments in groups of patients. As measures and therapies used during life-threatening conditions are often by themselves associated with serious adverse effects and complications, advanced monitoring is also necessary for the benefit of the individual patient. This experience was formulated long before the development of modern medicine: “Diseases desperate grown by desperate appliance are relieved, or not at all.” (Shakespeare, 1603; Hamlet).
REFERENCES

