


Akihiko Asahina, Yohei Minatani, Yayoi Tada, Hiroshi Mitsui and Kunihiko Tamaki
Department of Dermatology, Graduate School of Medicine, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8655, Japan. E-mail: asahina-tky@umin.ac.jp
Accepted June 9, 2005.
Sir,
Pyoderma gangrenosum (PG) is an uncommon, non-infectious, inflammatory skin disease characterized by progressive ulcer formation with undermined borders and a necrotic purulent base (1–4). It usually follows a chronic course, and there is no specific and uniformly effective treatment. Here, we present a patient with PG with recurrent ulcers on her extremities. Although her condition was refractory to conventional treatments, potassium iodide (KI), not routinely used in this disease, was found to be very effective in inducing remission.
CASE REPORT
A 35-year-old woman visited our hospital with multiple tender necrotic ulcers on her arms and legs. Laboratory examinations showed no abnormal findings except for a slight increase in C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR). A skin biopsy taken from the margin of a leg ulcer showed oedema and massive infiltration of neutrophils surrounded by lymphocytes and histiocytes in the dermis, and necrotic epidermis with hyperplastic change. There was no evidence of vasculitis. Tissue cultures did not reveal any pathogenic microorganisms. She had neither systemic involvement nor accompanying complications such as arthritis, inflammatory bowel disease, haematological disorders or malignancy. A diagnosis of classical type idiopathic PG was made.
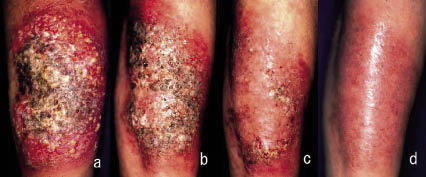
During the following 10 years, the ulcers constantly recurred on her extremities despite systemic treatments. This time, while continuing to take prednisolone 17.5 mg/day together with dapsone 50 mg/day and minocycline 200 mg/day as an adjuvant therapy, a well-demarcated new ulcer emerged and extended with marginal pustules following a minor injury on her left lower arm (Fig. 1a), and new lesions developed on both legs as well in 4 months. Instead of increasing the dose of steroid, we started oral KI at 900 mg/day. Surprisingly, the formation of new pustules almost stopped within a few days and improvement of the ulcers was observed accordingly (Fig. 1b and c). The dose of KI was increased to 1200 mg/day after 2 weeks, while the ulcers continued to regress rapidly. They disappeared almost completely after 1 month of KI therapy (Fig. 1d). The patient had not experienced such rapid resolution with any other treatment.DISCUSSION
Fig. 1. Clinical pictures of the large ulcer on the left lower arm during treatment. A large ulcer was seen with overlying necrotic tissue and pustules. Treatment with oral potassium iodide induced rapid resolution of the ulcer. (a) Before KI treatment, (b) day 12, (c) day 25, and (d) day 38.
PG is often difficult to manage because of its recurrent nature and poor responsiveness to therapy. Although local wound care and topical agents may be sufficient to control the disease process in mild cases, a combination with systemic therapy is usually necessary (1–4). For systemic therapy, oral corticosteroids offer the best results and remain the mainstay of treatment. Dapsone and minocycline are most frequently subscribed as steroid-sparing agents or as useful monotherapies in less aggressive cases. Immunosuppressive agents have been suggested in patients refractory to other therapies or who develop steroid-related side effects (2). They include azathioprine, cyclophosphamide, chlorambucil, cyclosporine, tacrolimus and mycophenolate mofetil. Combination therapies or multi-step approaches are common. However, many of these agents provide either incomplete long-term control of the disease or have been associated with adverse effects after chronic administration (5). In addition, there are no established guidelines, and no randomized controlled trials have been performed to assess their effectiveness (2, 4).
Our patient was treated with simultaneous administration of oral prednisolone, dapsone and minocycline, but they were not very helpful in controlling the exacerbation of disease. However, the effect of KI was so remarkable as to induce almost complete healing of ulcers within a month. KI is a traditional drug used for more than 150 years, and it continues to be used for sporotrichosis in most parts of the world (6). The use of KI was repopularized by the report of its effectiveness in erythema nodosum and nodular vasculitis (7). Horio et al. (8) found its usefulness for other inflammatory dermatoses, such as Behçet’s disease, erythema multiforme and Sweet’s syndrome. As for PG, Sanburg & Benzie (9) described a patient with Crohn’s disease and PG whose skin lesions healed after KI treatment. Richardson & Callen (10) also showed its effectiveness for recalcitrant PG. However, no additional cases have been reported except for these isolated cases.
The precise mechanism by which KI exerts its therapeutic effect against inflammatory dermatoses remains unknown (6). Neutrophil chemotaxis (11) and ability to generate the toxic oxygen intermediates (12) are suppressed by KI in vitro, and its pharmacological effect on neutrophils may be assumed. In this sense, dapsone and minocycline are also known to modify neutrophil function. The effectiveness of colchicine on PG (5) can be ascribed to the same mechanism in addition to its immunomodulatory effects. Prescription of KI is now established for Sweet’s syndrome which is characterized by an intense infiltration of neutrophils in the dermis (8, 13). Although the typical forms of PG and Sweet’s syndrome are clinically quite distinct and unlikely to be confused (13), the two conditions are among the same category of neutrophilic dermatoses. Interestingly, both PG and Sweet’s syndrome occur in the same systemic disease setting, such as malignancy, particularly haematological disorders. Both conditions show pathergic phenomena, similar response to immunosuppressive agents, and are known to develop after granulocyte-colony stimulating factor administration. Atypical or bullous PG is sometimes difficult to distinguish from Sweet’s syndrome. Histologically, bullous PG and early lesion of PG may closely mimic Sweet’s syndrome. Patients with both conditions concurrently have been described, even without an associated disease. Taken together these observations indicate the overlapping clinical spectrum and pathogenesis of PG and Sweet’s syndrome as an inflammatory process mediated by neutrophils (2, 3, 13). The usefulness of KI in our case of PG further supports this concept. Similar to our patient, Smith et al. (14) reported a case of Sweet’s syndrome in which KI was tried successfully after oral prednisolone and sequential addition of dapsone and minocycline all failed to produce clinical benefit. Interestingly, because their patient showed poor infiltration of neutrophils histologically, the authors speculated that KI might have an influence on the immune system rather than simply an effect on neutrophil function. Indeed, both PG and Sweet’s syndrome patients seem to have defective immune responses (13).
Nowadays, other treatment options have been advocated for PG (1), such as intravenous immunoglobulin and anti-TNF-α antibodies. Yet, KI can be used at very low cost and has only minor adverse effects. Although iodide-induced hypothyroidism can occur rarely as a result of defective autoregulation, it is usually reversible and returns to normal after stopping KI treatment (6). Curiously, the individual responsiveness to KI seems to vary; KI may even induce flare up of the disease in some patients with PG (2, 4). More clinical studies are necessary to elucidate which type of PG will respond well to KI.
In conclusion, our experience of dramatic effectiveness of KI for PG suggests common pathogenesis between PG and Sweet’s syndrome. KI should be an alternative choice for treatment of PG, at least when other treatments fail, are contraindicated, or cause intolerable side effects (6).
REFERENCES
1. Ehling A, Karrer S, Klebl F, Schaffler A, Muller-Ladner U. Therapeutic management of pyoderma gangrenosum. Arthritis Rheum 2004; 50: 3076–3084.
2. Callen JP. Pyoderma gangrenosum. Lancet 1998; 351: 581–585.
3. Crowson AN, Mihm MC Jr, Magro C. Pyoderma gangrenosum: a review. J Cutan Pathol 2003; 30: 97–107.
4. Chow RK, Ho VC. Treatment of pyoderma gangrenosum. J Am Acad Dermatol 1996; 34: 1047–1060.
5. Kontochristopoulos GJ, Stavropoulos PG, Gregoriou S, Zakopoulou N. Treatment of pyoderma gangrenosum with low-dose colchicine. Dermatology 2004; 209: 233–236.
6. Sterling JB, Heymann WR. Potassium iodide in dermatology: a 19th century drug for the 21st century – uses, pharmacology, adverse effects, and contraindications. J Am Acad Dermatol 2000; 43: 691–697.
7. Schulz EJ, Whiting DA. Treatment of erythema nodosum and nodular vasculitis with potassium iodide. Br J Dermatol 1976; 94: 75–78.
8. Horio T, Danno K, Okamoto H, Miyachi Y, Imamura S. Potassium iodide in erythema nodosum and other erythematous dermatoses. J Am Acad Dermatol 1983; 9: 77–81.
9. Sanburg AL, Benzie JL. An unusual use for an old drug: potassium iodide in pyoderma gangrenosum. Aust J Hosp Pharm 1987; 17: 187–188.
10. Richardson JB, Callen JP. Pyoderma gangrenosum treated successfully with potassium iodide. J Am Acad Dermatol 1993; 28: 1005–1007.
11. Honma K, Saga K, Onodera H, Takahashi M. Potassium iodide inhibits neutrophil chemotaxis. Acta Derm Venereol 1990; 70: 247–249.
12. Miyachi Y, Niwa Y. Effects of potassium iodide, colchicine and dapsone on the generation of polymorphonuclear leukocyte-derived oxygen intermediates. Br J Dermatol 1982; 107: 209–214.
13. Lear JT, Atherton MT, Byrne JP. Neutrophilic dermatoses: pyoderma gangrenosum and Sweet’s syndrome. Postgrad Med J 1997; 73: 65–68.
14. Smith HR, Ashton RE, Beer TW, Theaker JM. Neutrophil-poor Sweet’s syndrome with response to potassium iodide. Br J Dermatol 1998; 139: 555–556.

