


Yanxia Hou1,2, Jianjun Chen1,2, Min Gao1,2, Fusheng Zhou 2, Wenhui Du1,2, Yujun Shen2, Sen Yang1,2 and Xue-Jun Zhang1,2
1Institute of Dermatology & Department of Dermatology, First Hospital, Anhui Medical University, Hefei, and 2Key Laboratory of Genome Research, Anhui, Hefei, China
Dyschromatosis symmetrica hereditaria (OMIM127400) is a rare autosomal dominant pigmentary genodermatosis caused by mutations in the RNA-specific adenosine deaminase (ADAR) gene. This study investigated 5 families and 3 sporadic patients with dyschromatosis symmetrica hereditaria in the Chinese Han population from Anhui province, China. By direct sequencing, 5 novel ADAR gene mutations (c.982C > T, c.1491insA, c.2568_2571delTAAC, c.2969C > G and c.3040G > T) and 3 mutations described previously (c.3203-2A > G, c.3247C > T and c.3286C > T) were identified, all of which were heterozygous. We reviewed a total of 48 mutations in the ADAR gene in patients with dyschromatosis symmetrica hereditaria by previous reports and speculated that the mutation hotspots on the ADAR gene might be located in exons 9–15. The tRNA-specific and double-stranded RNA adenosine deaminase domain is essential for the deaminase activity of the ADAR encoded protein. Key words: ADAR gene; dyschromatosis symmetrica hereditaria; mutation.
(Accepted July 25, 2006.)
Acta Derm Venereol 2007; 87: 18–21.
Xue-Jun Zhang, Institute of Dermatology, Anhui Medical University, 69 Meishan Road, Hefei, Anhui 230022, China. E-mail: ayzxj@vip.sina.com
Dyschromatosis symmetrica hereditaria (DSH, OMIM127400), also called “reticulate acropigmentation of Dohi” or “symmetric dyschromatosis of the extremities”, was first described by Toyama in a Japanese family and was coined DSH in 1929 (1–3). This condition is a rare pigmentary genodermatosis characterized by a mixture of hyperpigmented and hypopigmented macules of various sizes, mostly on the dorsal aspects of the extremities. On the face, the lesions resemble ephelides and no hypopigmentation appears. These abnormalities are otherwise asymptomatic and do not affect the general health of the patient. The onset of the disease is usually during infancy or early childhood. Recently, Zhang et al. (4) found the age of disease onset could be as high as 15 years. DSH has been reported mainly in Japan and China, although it occurs in families of all ethnic origins worldwide (5). Autosomal dominant and recessive inheritance patterns as well as sporadic cases of DSH have all been described (5, 6).
The genetic basis of DSH has been elucidated recently. Zhang et al. (7) mapped the DSH gene on chromosome 1q11–1q21. With the identification of mutations in the RNA-specific adenosine deaminase gene (ADAR, OMIM 601059) among Japanese families with DSH, Miyamura et al. confirmed that the ADAR gene is responsible for DSH (8). Up to now, a total of 43 ADAR gene mutations have been reported in patients with DSH (4, 8–20). In this study, mutation scanning was performed by direct sequencing of polymerase chain reaction (PCR) products derived from genomic DNAs in 5 Chinese families and 3 sporadic patients with DSH. Eight ADAR mutations were found, all of which were heterozygous. These mutations may be useful for expanding the database of ADAR mutation and investigating the still-unknown mechanism leading to DSH.
MATERIALS AND METHODS
Patients
This study investigated 5 families (Family 1–5) (Fig. 1) and 3 sporadic cases (Patient 1–3) with DSH in the Chinese Han population from Anhui province. Five multi-generation DSH families exhibited autosomal dominant inheritance. All affected individuals had a typical mixture of hyperpigmented and hypopigmented macules on the extremities. These lesions were irregular shapes and sizes. The age of onset varied from 3 months to 8 years. In Families 2 and 3, the affected individuals had characteristic macules around the elbow and knee joints and small freckle-like pigmented macules on the face. In Family 2 and Patient 2, the lesions may become aggravated upon prolonged sun exposure in the summer. DSH was diagnosed by experienced dermatologists based on the typical manifestations and histopathological findings.
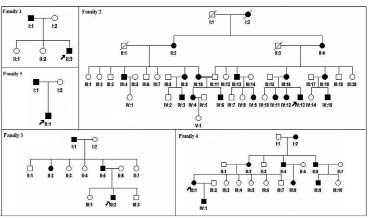
Fig. 1. Pedigrees of the 5 families with dyschromatosis symmetrica hereditaria. Affected family members are represented by black symbols. The probands are arrowed.
Mutation analysis
The study was approved by the Anhui Medical University Review Board and all study subjects gave informed consent. Peripheral blood samples were taken from all the available affected and unaffected individuals. Genomic DNAs were extracted from the blood samples using a blood kit (Promega, Madison, Wi, USA). In addition, genomic DNA samples were extracted from 100 unrelated healthy volunteers as controls to exclude the polymorphisms in the ADAR gene. The primers that flanked all 15 coding exons and intron-exon boundaries of the ADAR gene were designed using the web-based version of the Primer 3.0 program (http://www.genome.wi.mit.edu/ cgi-bin/primer/primer3_www.cgi). All exons of the ADAR gene were amplified by PCR. PCR was performed in 20-µl reaction volume containing 20 ng of genomic DNA, 0.3 mM dNTPs, 0.3 µM of each primer, 3.0 mM MgCl2 and 0.1 U of Taq DNA polymerase. The PCR conditions were: Taq activation at 95°C for 5 min, followed by 35 cycles consisting of denaturation at 94°C for 40 s, annealing at 58°C for 60 s and extension at 72°C for 55 s, and the final extension was 72°C for 10 min. After the amplification, the products were purified on a 1.5% agarose gel. We directly sequenced ADAR gene on ABI PRISM 3730 automated sequencer (Applied Biosystems). Sequence comparisons and analysis were performed using Phred-Phrap-Consed program, version 12.0. We describe amino acid position in ADAR according to ADAR GenBank sequence: NM_001111.
RESULTS
Among 5 Chinese families and 3 sporadic patients with DSH, we detected 5 novel ADAR mutations and 3 previously described mutations by direct sequence analysis of the PCR products. The spectrum of mutations included 2 missense and 3 nonsense mutations, 2 frameshift mutations and 1 splice acceptor mutation (Fig. 2).
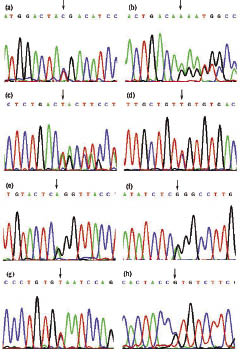
Fig. 2. Mutations of the RNA-specific adenosine deaminase gene found in families and sporadic patients with dyschromatosis symmetrica hereditaria:
(a) c.3286C > T (p.R1096X) mutation in Family 1.
(b) c.1491insA (p. T497fs) mutation in Family 2.
(c) c.2568_2571delTAAC (p.T856fs) mutation in Family 3.
(d) c.3247C > T (p.R1083C) mutation in Family 4.
(e) c.3203-2A > G mutation in Family 5.
(f) c.982C > T (p. R328X) mutation in Patient 1. Antisense sequence is shown in this sequence.
(g) c.3040G > T (p.E1014X) mutation in Patient 2.
(h) c.2969C > G (p.P990R) mutation in Patient 3.
The c.982C > T (p. R328X) mutation was identified in Patient 1, but not in his healthy parents. The predicted protein lacked 899 amino acids. The nonsense mutation c.3040G > T (p. E1014X) in exon 12 was detected in Patient 2. The predicted protein, with a defective ADEAMc domain, lacks 213 amino acids. The proband and his father in Family 1 had the c.3286C > T (p. R1096X) mutation in exon 13, leading to a truncated protein that lacks 131 amino acids in the putative deaminase domain. This mutation was reported previously by Zhang et al. (4).
We detected a frameshift mutation in Family 2. The nucleotide A was inserted between the 1491 and 1492 nucleotide, which results in the mutation of c.1491insA (p.T497fs→516X). This mutation was found in exon 2 and confirmed in the other patients. The other frameshift mutation (p. T856fs) was found in all patients from Family 3. The 4 nucleotides TAAC were deleted at nucleotides from 2568 to 2571, which led to frameshift and premature translation termination in exon 8. The truncated protein lacked the putative deaminase domain.
A missense mutation c.2969C > G (p. P990R) in exon 11 was carried by Patient 3. This transition located in the highly conserved region of the deaminase domain changes codon 990 from proline (CCT) to arginine (CGT). A C→T transition was found at nucleotide 3247 of ADAR gene in Family 4. This transition replaced a highly conserved arginine residue with cysteine in codon 1083. The amino acid residue was located in the ADEAMc domain. This mutation was reported previously by Sun et al. (16). In Family 5, a c.3203-2A > G mutation alters the splice acceptor sequence of IVS12 and should prevent proper splicing of the transcript. This mutation was also reported previously by Zhang et al. (4).
DISCUSSION
DSH is a rare hereditary skin disease characterized by a mixture of hyperpigmented and hypopigmented macules of various sizes on the backs of the hands and feet. Miyamura et al. (8) firstly identified mutations in the ADAR gene responsible for DSH. ADAR, also called DSRAD (double-stranded RNA-specific adenosine deaminase), belongs to a family of RNA-specific adenosine deaminase that represents one type of RNA editing enzyme. ADAR catalyses the deamination of adenosine in regions of dsRNA to form inosine (A to I editing), which is read-out at the ribosome level as guanosine. The enzyme is found only in vertebrates, but detected ubiquitously in different tissues (21, 22). The gene spans 30 kb and contains 15 exons and its encoded protein is composed of 1226 amino acid residues (22, 23). Two Z-alpha motifs and three dsRNA-binding motif (DRBM) repeats spread over exon 2 and exons 2–7, respectively. The ADEAMc domain encompasses exons 9–14 (22). Each DBRM domain of ADAR could enhance the efficiency of editing and the ADEAMc domain is essential for editing (21). Through the selective modification of both codons and splice sites by ADAR enzyme, a number of different protein products can be generated from the same gene (24). Abnormal RNA editing may induce the differentiation of melanoblasts to hyperactive or hypoactive melanocytes, then colonizing in an irregular distribution in the skin lesions.
In this study, we detected 2 missense and 3 nonsense mutations, 2 frameshift mutations and 1 splice acceptor mutation among 5 families and 3 sporadic cases with DSH. The nonsense mutation p.R328X and the frameshift mutation p.T497fs→516X could cause an ADAR protein truncation lacking three dsRNA-binding motifs and a catalytic domain. It is possible that the truncated proteins without functional activity could underlie the pathogenesis of DSH. The other 2 nonsense mutations (p. E1014X, p. R1096X) and the frameshift mutation (p. T856fs) may induce premature translation termination of the protein, which results in defective deaminase activity. The transition C→G at nucleotide 2969 in the highly conserved region of the deaminase domain may change codon 990 from proline (CCT) to arginine (CGT). The C→T mutation at nucleotide 3247 of ADAR gene replaced a highly conserved arginine residue with cysteine in codon 1083. The amino acid residue was located in the ADEAMc domain. Both of the DRBMs and the ADEAMc domain were required for formation of the homodimer. The homodimer is essential for the enzyme activity of ADAR protein (24). The mutation might prevent the formation of the homodimer by a different amino acid and change the catalytic activity of the enzyme. The c.3203-2A > G mutation should lead to improper splicing of the transcript and lead to the change of conformation and activity of the enzyme.
We reviewed all the mutation reports of ADAR gene and found that most of the mutations occur in exons 9–15 (Fig. 3). The ADEAMc domain is very important for the deaminase activity of the ADAR protein. Exon 15 is not involved in encoding the catalytic domain and how to explain the mutations in this exon. Gao et al. (13) speculated that these mutations will change the structure of the enzyme and may induce an unstable tertiary structure of the protein that results in defective activity. Liu et al. (20) found that all the missense mutations are restricted to the catalytic domain. We compared the clinical features with the mutation identified in all families, but did not find a clear correlation between genotypes and phenotypes. The same mutation will lead to different phenotypes even in the same family. Suzuki et al. (15) and Liu et al. (20) state that DSH might be caused by haploinsufficiency of the ADAR activity. Although the ADAR gene is expressed ubiquitously in different tissues and the role of ADAR protein in diversifying codons is important, the reason why the pigmentary changes in DSH are localized specifically on the backs of the hands and feet is unknown.
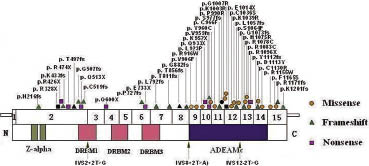
Fig. 3. Map of functional domains of RNA-specific adenosine deaminase gene mutations responsible for dyschromatosis symmetrica hereditaria. The exons of the ADAR cDNA are numbered with Arabic numerals. The 3 functional domains (Z-alpha, DRBMs, ADEAMc) are indicated in the figure by different colours. The new mutations identified in this study are indicated by black symbols. Previously reported mutations are indicated by orange, green and purple symbols, respectively. Black arrows indicate mutations in splice acceptor sequences.
In conclusion, we have reported 5 novel mutations of ADAR involved in DSH. The establishment of a mutation spectrum of the ADAR gene may be useful for investigating the mechanism leading to DSH. The role of the ADAR protein in differentiation of melanoblasts requires further clarification.
ACKNOWLEDGEMENTS
We are most grateful to all members of the DSH families for taking part in our study. This work was funded by grants from the Chinese High-Tech Program (863) (2003AA227030).
REFERENCES
1. Ostlere LS, Ratnavel RC, Lawlor F, Black MM, Griffiths WA. Reticulate acropigmentation of Dohi. Clin Exp Dermatol 1995; 20: 477–479.
2. Toyama I. An unknown disorder of hyperpigmentation. Jpn J Dermatol Urol 1910; 10: 644.
3. Toyama I. Dyschromatosis symmetrica hereditaria. Jpn J Dermatol Urol 1929; 29: 95–96.
4. Zhang XJ, He PP, Li M, He CD, Yan KL, Cui Y, et al. Seven novel mutations of the ADAR gene in Chinese families and sporadic patients with dyschromatosis symmetrica hereditaria (DSH). Hum Mutat 2004; 23: 629–630.
5. Oyama M, Shimizu H, Ohata Y, Tajima S, Nishikawa T. Dyschromatosis symmetrica hereditaria (reticulate acropigmentation of Dohi): report of a Japanese family with the condition and a literature review of 185 cases. Br J Dermatol 1999; 140: 491–496.
6. Alfadley A, Al Ajlan A, Hainau B, Pedersen KT, Al Hoqail I. Reticulate acropigmentation of Dohi: a case report of autosomal recessive inheritance. J Am Acad Derm 2000; 43: 113–117.
7. Zhang XJ, Gao M, Li M, Li CR, Cui Y, He PP, et al. Identification of a locus for dyschromatosis symmetrica hereditaria at chromosome 1q11–1q21. J Invest Dermatol 2003; 120: 776–780.
8. Miyamura Y, Suzuki T, Kono M, Inagaki K, Ito S, Suzuki N, et al. Mutations of the RNA-specific adenosine deaminase gene (ADAR) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet 2003; 73: 693–699.
9. Liu Q, Liu W, Jiang L, Sun M, Ao Y, Zhao X, et al. Novel mutations of the RNA-specific adenosine deaminase gene (ADAR) in Chinese families with dyschromatosis symmetrica hereditaria. J Invest Dermatol 2004; 122:896–899.
10. Li M, Jiang YX, Liu JB, Yang S, He PP, Gao M, et al. A novel mutation of the ADAR gene in a Chinese family with Dyschromatosis symmetrica hereditaria. Clin Exp Dermatol 2004; 29: 533–535.
11. Yang Y, Li S, Li H, Bu DF, Wang K, Tu P, et al. ADAR gene mutations in three families with dyschromatosis symmetrica hereditaria. Beijing Da Xue Xue Bao 2004; 36: 466–468 [in Chinese].
12. Li CR, Li M, Ma HJ, Luo D, Yang LJ, Wang DG, et al. A new arginine substitution mutation of ADAR gene in a Chinese family with dyschromatosis symmetrica hereditaria. J Dermatol Sci 2005; 37: 95–99.
13. Gao M, Wang PG, Yang S, Hu XL, Zhang KY, Zhu YG, et al. Two frameshift mutations in the RNA-specific adenosine deaminase gene associated with dyschromatosis symmetrica hereditaria. Arch Dermatol 2005; 141: 193–196.
14. Cui Y, Wang J, Yang S, Gao M, Chen JJ, Yan KL, et al. Identification of a novel mutation in the ADAR gene in a Chinese pedigree with dyschromatosis symmetrica hereditaria. Arch Dermatol Res 2005; 296: 543–545.
15. Suzuki N, Suzuki T, Inagaki K, Ito S, Kono M, Fukai K, et al. Mutation analysis of the ADAR1 gene in dyschromatosis symmetrica hereditaria and genetic differentiation from both dyschromatosis universalis hereditaria and acropigmentatio reticularis. J Invest Dermatol 2005; 124: 1186–1192.
16. Sun XK, Xu AE, Chen JF, Tang X. The double-RNA-specific adenosine deaminase (ADAR) gene in dyschromatosis symmetrica hereditaria patients: two novel mutations and one previously described. Br J Dermatol 2005; 153: 342–345.
17. Xing Q, Wang M, Chen X, Qian X, Qin W, Gao J, et al. Identification of a novel ADAR mutation in a Chinese family with dyschromatosis symmetrica hereditaria (DSH). Arch Dermatol Res 2005; 297: 139–142.
18. Li M, Li C, Hua H, Zhu W, Lu Y, Yang L. Identification of two novel mutations in Chinese patients with Dyschromatosis symmetrica hereditaria. Arch Dermatol Res 2005; 8: 1–5.
19. Chao SC, Lee JY, Sheu HM, Yang MH. A novel deletion mutation of the ADAR gene in a Taiwanese patient with dyschromatosis symmetrica hereditaria. Br J Dermatol 2005; 153: 1064–1066.
20. Liu Q, Jiang L, Liu WL, Kang XJ, Ao Y, Sun M, et al. Two novel mutations and evidence for haploinsufficiency of the ADAR gene in dyschromatosis symmetrica hereditaria. Br J Dermatol 2006; 154: 636–642.
21. Herbert A, Wagner S, Nickerson JA. Induction of protein translation by ADAR1 within living cell nuclei is not dependent on RNA editing. Mol Cell 2002; 10: 1235–1246.
22. Wang Y, Zeng Y, Murray JM, Nishikura K. Genomic organization and chromosomal location of the human dsRNA adenosine deaminase gene: the enzyme for glutamate-activated ion channel RNA editing. J Mol Biol 1995; 254: 184–195.
23. O’Connell MA, Krause S, Higuchi M, Hsuan JJ, Totty NF, Jenny A, et al. Cloning of cDNAs encoding mammalian double-stranded RNA-specific adenosine deaminase. Mol Cell Biol 1995; 15: 5376–5388.
24. Cho DS, Yang W, Lee JT, Shiekhattar R, Murray JM, Nishikura K. Requirement of dimerization for RNA editing activity of adenosine deaminases acting on RNA. J Biol Chem 2003; 278: 17093–17102.

