Sir,
Antiphospholipid syndrome (APS) is characterized by thromboses, cutaneous manifestations, recurrent foetal loss and thrombocytopenia, which appear concomitantly with antiphospholipid autoantibodies (1). Laboratory tests should reveal either persistent lupus anticoagulant (LAC) or anticardiolipin antibodies (ACA) (2). Secondary APS appears together with systemic lupus erythematosus, vasculitic disorders, infections, medication or malignancies. More often, APS is primary without any causative reason. Cutaneous manifestations result from generalized coagulopathy and play a pivotal role in the clinical picture (3). These include livedo reticularis, livedoid vasculitis, thrombophlebitis, acrocyanosis, atrophie blanche, Raynaud’s phenomenon, cutaneous necrosis, erythematous macules, purpura, painful nodules, subungual splinter haemorrhages and scleroderma-associated cutaneous signs. Histopathology usually reveals non-inflammatory vasculopathy, thrombosis, endothelial damage, haemorrhagic signs, haemosiderin, oedema and necrosis (4). While perimyocarditis, endocarditis and pericardial effusion may develop in connective tissue diseases (5–7), APS-associated cardiac complications typically include valvular heart disease, valve vegetations, intracardiac thrombus and coronary artery disease (8). In primary APS cardiac effusion is extremely rare and only a few cases of tamponade have been reported in French literature (9, 10). In this report, we describe a patient who had primary APS with skin and eye complications only, who eventually developed massive pericardial effusion and succumbed to sudden cardiac tamponade.
CASE REPORT
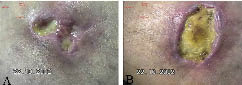
A 47-year-old male bus driver with no particular previous medical history developed painful necrotic ulcerations above both medial malleoli (Fig. 1). He had suffered from Raynaud’s phenomenon in his hands. Upon admittance, he had subungual splinter haemorrhages and porcelain-white scars on the arms. Nodules and crusts had been present on the dorsal aspects of his hands and fingers for a few months. Histopathology of the ulcers first pointed to vasculitic features in a medium-sized artery, suggesting cutaneous polyarteritis nodosa. Thromboses were not evident in two distinct biopsies. Angiography excluded atherosclerosis and provided no evidence of systemic polyarteritis in the lower extremities or kidneys.
Fig. 1. Following 3 months of admission, the medial malleoli-associated leg ulcers on the right (A) and left (B) extremity were painful, fibrin-rich, devoid of healthy granulation tissue and had actively inflamed edges. Atrophie blanche-like white-coloured stellate scars developed subsequently.
White blood cell count, haemoglobin, platelet count, C-reactive protein, erythrocyte sedimentation rate, plasma glucose and urine samples were normal. Thyroid, renal and liver function tests had no major abnormalities. Fluvastatine (20 mg) normalized plasma total cholesterol from 7.3 to 5.1 mmol/l. Immunological tests for antinuclear antibodies, extractable nuclear antigen antibodies (including SCL-70), serum/plasma cryoglobulins and anti-neutrophil cytoplasmic antibodies including proteinase-3 and myeloperoxidase antibodies were negative. Circulating immune complexes were not detected in the PIPA-test (platelet 125I-labelled staphylococcal protein A test). Immunofluorescence assays could not detect specific antibodies against native DNA, mitochondria or centromeres. Lupus anticoagulant (LAC) was positive 1.38 (<1.23) with the PTT-LA (LAC-sensitive activated partial thromboplastin time) test. LAC was first normal 1.04 (<1.08) with the dilute Russell viper venom test (dRVVT). Twelve weeks later, control analysis yielded LAC in both tests (PTT-LA 1.25 and dRVVT 1.09) suggesting APS. Repeated tests for anti-cardiolipin and beta-2-glycoprotein antibodies were negative. Thromboplastin and prothrombin times, protein S and C as well as anti-thrombin III activities were normal. Factor V and prothrombin genes were not mutated.
HIV, hepatitis C virus, Treponema pallidum and Borrelia burgdorferi antibodies were not detected. Hepatitis B virus surface antigen, Chlamydia trachomatis nucleic acid tests and studies of gonococcal infection were also negative. Class G beta-haemolytic streptococci were cultured from the leg ulcers and penicillin was administered orally. Later, dental examination revealed periodontitis and a focus of dental abscess. Ceftriaxone was started for 2 days and continued with ciprofloxacin for another 5 days. Finally, a 3-week course of cefadroxil was administered to ensure eradication of dental and other putative infections. Chest X-ray revealed traces of marginal unilateral basal interstitial fibrosis.
Serum protein electrophoresis detected two bands in the gamma region. M-component was also present in the urine and immunoelectrophoresis (serum/urine) identified IgG class free kappa light-chains. Serum IgG level was increased to 20.2 g/l (range 6.77–15 g/l). Both IgM and IgA levels were normal, whereas IgE was increased to 1092 kU/l (< 110). Only 0.1 g/l of protein was found in urine. Bone marrow aspirate gave no evidence of myeloma, although gammopathy was suggested due to slightly increased plasma cell counts.
The ulcers remained resistant to topical therapies. The patient could not tolerate compression therapy because the ulcers were painful. Prior to the APS diagnosis, prednisolone (30–60 mg/day) was administered without response for several months. Methotrexate (10 mg oral dose/week), which was combined with the medication for 6 weeks, had no effect. A 3-month course of cyclosporine (200 mg/day) gave no response. After the repeatedly positive LAC and the suspicion of primary APS, combination of acetylsalicylic acid (25 mg) and dipyridamol (200 mg) was initiated twice a day. To further improve circulation, nifedipine (20 mg) and fluvastatine (20 mg) were added to the therapy. During this treatment the ulcers started to granulate and decrease in size, and were completely healed in 3 months. Later, the patient discontinued nifedipine and fluvastatine, which was followed by the development of new ulcerations. Nifedipine was re-added to the anti-aggregant therapy, and again the clinical condition improved.
The patient began to complain of disturbances in the vision of the right eye. Ophthalmological examination disclosed retinal thromboses. Later, fluorescein angiography revealed ischaemic changes. Despite repeated argon laser photocoagulation, vision was lost as a result of neovascularization glaucoma. To prevent thromboses in the left eye, acetylsalicylic acid was changed to warfarin.
After 3 years, the patient had developed scleroderma-like facial features and the fingers showed fibrotic hardening resembling sclerodactyly. No other symptoms or serological signs of systemic sclerosis were present, and the cutaneous signs of primary APS were constantly the predominant clinical feature. The patient discontinued warfarin and was hospitalized one month later due to infected necrotic leg ulcers. On the day of admittance he suddenly developed asphyxia and died despite resuscitation.
Autopsy revealed massive pericardial effusion (1480 g) that had caused cardiac tamponade. Thromboembolic complications were not found in pulmonary or coronary arteries or any other sites examined. Neither macroscopic atherosclerosis nor infarctions were found, and heart valves were normal. The heart was hypertrophic (520 g), pulmonary tissue contained mild fibrosis and there was clear and non-purulent pleural effusion (2000 ml). There was blood stasis in the liver and spleen that pointed to both acute and chronic cardiac insufficiency. Importantly, myocarditis or pericarditis was not found. Histopathology of the leg ulcers revealed thromboses of dermal vessels with no signs of vasculitis. The immediate cause of death was cardiac tamponade, which had developed without any other evident reason than the primary APS.
DISCUSSION
It has been suggested that cutaneous signs precede other clinical manifestations in 41% of patients with APS (11). The present case further demonstrates that they may, in addition, represent the predominant clinical picture, as thrombotic and other typical complications were not detected. The anti-aggregant therapy, however, lead to clear clinical improvement, thus further confirming the diagnosis of primary APS. According to the updated APS diagnosis criteria the patient should have at least one clinical (vascular thrombosis or pregnancy morbidity) and one laboratory finding (persistent LAC, ACA or anti-beta2-glycoprotein I antibodies) (12). Our patient had repeatedly positive LAC and he developed retinal thromboses. During the whole follow-up period the most evident diagnosis was primary APS and nothing pointed to systemic lupus erythematosus or any other autoimmune disorder. Still, it is possible that the patient was gradually developing a variant of scleroderma or other collagenosis-like disorder. Gammopathy was detected, but no evidence of myeloma or any other malignancy was obtained during the follow-up period or in the autopsy.
Antiphospholipid antibodies and increased coagulation have been reported to associate with infective endocarditis (13). In our case, there were no signs of peri- or endo-carditis. The pericardial effusion had likely developed slowly, as the liver and spleen had marked blood congestion indicative of chronic heart failure.
REFERENCES
1. Nahass GT. Antiphospholipid antibodies and the antiphospholipid antibody syndrome. J Am Acad Dermatol 1997; 36: 149–168.
2. McNeil HP, Chesterman CN, Krilis SA. Immunology and clinical importance of antiphospholipid antibodies. Adv Immunol 1991; 49: 193–280.
3. Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol 1997; 36: 970–982.
4. Alegre VA, Winkelmann RK. Histopathologic and immunofluorescence study of skin lesions associated with circulating lupus anticoagulant. J Am Acad Dermatol 1988; 19: 117–124.
5. Ferri C, Giuggioli D, Sebastiani M, Colaci M, Emdin M. Heart involvement and systemic sclerosis. Lupus 2005; 14: 702–707.
6. Doria A, Iaccarino L, Sarzi-Puttini P, Atzeni F, Turriel M, Petri M. Cardiac involvement in systemic lupus erythematosus. Lupus 2005; 14: 683–686.
7. Langley RL, Treadwell EL. Cardiac tamponade and pericardial disorders in connective tissue diseases: case report and literature review. J Natl Med Assoc 1994; 86: 149–153.
8. Tenedios F, Erkan D, Lockshin MD. Cardiac involvement in the antiphospholipid syndrome. Lupus 2005; 14: 691–696.
9. Bennis A, Nafidi S, Zahraoui M, Tahiri A, Chraibi N. Tamponnade cardiaque et accident ischémique transitoire révélant un syndrome des anticorps antiphospholipides primitif. Ann Cardiol Angeiol (Paris) 1998; 47: 19–21.
10. Noureddine M, Bennis A, Raquim S, Tahiri A, Chraibi N. Anomalies cardiovasculaires es révélatrices du syndrome des anticorps anti-phospholipides. Arch Mal Coeur Vaiss 2003; 96: 324–331.
11. Alegre VA, Gastineau DA, Winkelmann RK. Skin lesions associated with circulating lupus anticoagulant. Br J Dermatol 1989; 120: 419–429.
12. Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4: 295–306.
13. Kupferwasser LI, Hafner G, Mohr-Kahaly S, Erbel R, Meyer J, Darius H. The presence of infection-related antiphospholipid antibodies in infective endocarditis determines a major risk factor for embolic events. J Am Coll Cardiol 1999; 33: 1365–1371.