Epidermolysis bullosa (EB) pruriginosa is an unusual variant of dystrophic EB in which intense itching can lead to striking skin changes resembling acquired skin disorders such as nodular prurigo or hypertrophic lichen planus. The molecular pathology involves mutations in the COL7A1 gene, but the nature of the mutations is similar to those seen in other non-pruritic forms of dystrophic EB. The mechanism of the dramatic phenotypic differences is currently unknown. In this study we assessed the incidence of a common functional polymorphism in the matrix metalloproteinase-1 gene promoter (1G or 2G at nucleotide –1607) in individuals with EB pruriginosa (n = 27) compared with non-itchy dominant dystrophic EB (n = 23), recessive dystrophic EB (n = 25) and normal controls (n = 50). The hypothesis is that the 2G allele, which was previously shown to increase matrix metalloproteinase-1 activity and lead to increased degradation of type VII collagen, could explain the phenotypic heterogeneity encountered in dominant forms of EB, particularly the itchy EB pruriginosa phenotype. The rationale is that increased type VII collagen degradation could trigger an inflammatory response leading to itchy skin characteristic of EB pruriginosa. All 27 individuals with EB pruriginosa were heterozygous for dominant-negative glycine substitution mutations in the COL7A1 gene, six of which have not been reported previously. The frequency of the 2G allele in these subjects (46.3%) was greater than in the controls (42.0%), but less than in non-itchy dominant dystrophic EB (52.2%) or recessive dystrophic EB (62.0%), indicating that variants of a common functional polymorphism in the matrix metalloproteinase-1 gene promoter do not account for the itchy skin phenotype. The pathophysiology of EB pruriginosa remains unexplained. Key words: COL7A1; blister; inherited skin disease; pruritus.
(Accepted November 17, 2008.)
Acta Derm Venereol 2009; 89: 6–11.
John McGrath, St John’s Dermatology Research Laboratories, 9th Floor Tower Wing, Guy’s Hospital, Great Maze Pond, London SE1 9RT, UK. E-mail: john.mcgrath@kcl.ac.uk
All forms of dystrophic epidermolysis bullosa (DEB), both dominant and recessive, result from mutations in the COL7A1 gene, which encodes for type VII collagen, the major component of anchoring fibrils at the dermal-epidermal junction (1). Moreover, different combinations of nonsense, missense, frameshift or splice site mutations in COL7A1 have been shown to account for the clinical spectrum of DEB and a paradigm for genotype-phenotype correlation has been established (2, 3). Nevertheless, one particular subtype of DEB that has been difficult to categorize is epidermolysis bullosa pruriginosa (EBP) (OMIM 604129). This form of DEB, which was first reported in 1994 (4), shows some overlap with pretibial DEB (OMIM 131850), but the intense pruritus that occurs in EBP is striking and may lead to localized or generalized skin features resembling lichen simplex chronicus, nodular prurigo, immunobullous diseases, hypertrophic lichen planus or even dermatitis artefacta (4–6). The inheritance of EBP is usually autosomal dominant, but may be recessive in some cases (4, 6–8). Perhaps surprisingly, the nature of the pathogenic COL7A1 mutations does not appear to differ from other forms of DEB (5–7, 9–15). Indeed, there are several examples of identical mutations in individuals from the same or different families resulting in either EBP or a more classical form of DEB (5, 7, 10, 13–15).
In order to try to explain the phenotypic disparity, investigators have previously examined potential disease modifiers, including IgE levels, atopy, biochemical or endocrinological abnormalities, iron deficiency, and most recently, filaggrin mutations, but thus far no specific insight into the EBP phenotype has emerged (6–8, 11, 16, 17). Therefore, the significance of other metabolic, genetic, epigenetic, or environmental factors in contributing to the EBP phenotype is currently unknown.
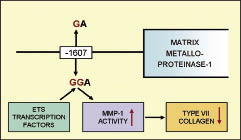
Recently, Titeux et al. (18) reported that a polymorphism within the promoter region of one of the major zinc metallo-endopeptidases genes, matrix metalloproteinase-1 (MMP1), is capable of increasing transcription of the enzyme two-fold. MMPs can degrade various extracellular molecules, including type VII collagen, and play a key role in tissue remodelling and wound healing (19–23). With regards to the MMP1 gene promoter, the sequence at the –1607 position can be either 1G or 2G, both of which have been shown to occur with similar frequency in control populations (22, 23). However, the 2G polymorphism creates a new ETS transcription factor binding site, which increases the transcriptional activity of MMP1, leading to increased degradation of type VII collagen (Fig. 1). The ETS family of transcription factors play important roles in development, differentiation and proliferation of cells in general and are involved in apoptosis and tissue remodelling. Titeux et al. (18) were able to show that the 2G polymorphism (on either one or both alleles) correlates with disease severity in individuals with recessive DEB and that type VII collagen levels at the dermal-epidermal junction are a more important clinical determinant than the amount of type VII collagen synthesis. Thus, this promoter polymorphism in the MMP1 gene has been shown to be a potential disease modifier in one variant of DEB. This raises the question as to whether clinical variation in other forms of DEB might also be explained by this mechanism. The aim of our study, therefore, was to assess whether this particular polymorphism might have relevance to the pathophysiology of EBP. The rationale is that increased degradation of type VII collagen could induce secondary inflammatory changes, including expression of cytokines and mediators known to be associated with skin itching (6, 7).
Fig. 1. The 1G/2G polymorphism in the matrix metalloproteinase 1 gene promoter is located at nucleotide –1607 from the MMP1 transcription site. The 2G polymorphism creates an ETS transcription factor binding site, which increases the transcriptional activity of MMP1; this leads to increased degradation of type VII collagen.
METHODS
Following ethical approval (St Thomas’ Hospital Ethics’ committee; 07/H0802/104) and informed consent, individuals with EBP (n = 27), non-EBP dominant DEB (n = 23), recessive DEB (n = 25) and normal control subjects (n = 50) were recruited for the study through the Robin Eady National Diagnostic EB Laboratory database in London. Each group comprised 70–80% white Caucasians, the remainder were of Middle-Eastern, South American, South-East Asian or Asian origins. There were no major differences between the ethnicity of any of the subject groups, including the controls. In subjects with EBP, clinical and laboratory investigations revealed no underlying cause(s) for the pruritus including atopy, iron deficiency, biochemical or endocrinological abnormalities, although none has yet been screened for mutations in the filaggrin (FLG) gene.
Genomic DNA was extracted from peripheral blood leukocytes using standard methods (24). COL7A1 mutation screening was performed by direct sequencing of all 118 exons and flanking introns of the COL7A1 gene, as described previously, using an ABI 3100 sequencer (25). Mutations were verified by bi-directional sequencing and restriction endonuclease digestions, where possible.
To sequence the MMP1 promoter region corresponding to the predicted 1G/2G polymorphism (–1607bp from the MMP1 transcription site), the following primers were used: forward primer 5’-gtggaagcttacacctataatcccaacactc-3’ (–4008 bp to –3988 bp; GenBank No NM002421) and reverse primer 5’-ctgcctggtaccctattgcgatagcaccatggc-3’ (–511 bp to –543 bp), with polymerase chain reaction (PCR) amplification conditions as described previously (26). The frequency of the two alleles was compared between the different groups using Fisher’s exact test.
RESULTS
All 27 subjects with EBP (19 families) had dominant DEB resulting from heterozygous glycine substitutions in the type VII collagen triple helix. Six of these mutations have not been reported previously (details are shown in Table I) and thus our mutation screening documents the COL7A1 molecular pathology in the largest cohort of individuals with EBP (Fig. 2). The COL7A1 mutations in the 23 non-EBP dominant individuals (20 families) comprised 13 heterozygous glycine substitutions, six unreported (Table I) and a new splice site mutation; IVS37+1G > T (Fig. 2). Of note, however, there were no major differences in the location of the glycine substitutions within the type VII collagen protein when comparing the EBP and non-EBP dominant DEB groups (Fig. 2). These cohorts also contained 12 individuals with the same COL7A1 mutation, p.G2043R, which is the most common mutation found in dominant DEB (27, 28). The phenotype for this particular glycine substitution was EBP in four cases and non-EBP dominant DEB in the other eight. Thus, the same mutation can underlie both clinical subtypes, as illustrated in Fig. 3.
Table I. Previously unreported COL7A1 mutations identified in this study
|
Mutation
|
cDNA sequence change
|
Position
|
Phenotype
|
|
IVS37+1G > T
|
IVS37+1G > T
|
Intron 37
|
DDEB
|
|
p.G1483D
|
c.4448 G > A
|
Exon 42
|
BDN
|
|
p.G1770D
|
c.5309 G > A
|
Exon 61
|
EBP
|
|
p.G1773D
|
c.5318 G > A
|
Exon 61
|
DDEB
|
|
p.G1860R
|
c.5578 G > A
|
Exon 66
|
EBP
|
|
p.G1913R
|
c.5737 G > C
|
Exon 69
|
EBP
|
|
p.G2009A
|
c.6026 G > C
|
Exon 73
|
DDEB
|
|
p.G2028E
|
c.6083 G > C
|
Exon 73
|
DDEB
|
|
p.G2067A
|
c.6200 G > C
|
Exon 74
|
DDEB
|
|
p.G2159E
|
c.6476 G > A
|
Exon 79
|
EBP
|
|
p.G2213R
|
c.6637 G > A
|
Exon 83
|
EBP
|
|
p.G2233E
|
c.6698 G > A
|
Exon 84
|
BDN
|
|
p.G2290A
|
c.6869 G > C
|
Exon 87
|
EBP
|
DDEB: dominant dystrophic epidermolysis bullosa; BDN: bullous dermolysis of the newborn; EBP: epidermolysis bullosa pruriginosa.
Fig. 2. COL7A1 mutation analysis in individuals with dominant dystrophic epidermolysis bullosa (DEB) in this study. The mutations in 27 cases of epidermolysis bullosa pruriginosa (EBP) (19 families) are shown at the top of the figure and the mutations in 23 cases of non-EBP dominant DEB (20 families) are shown below. The white boxes with blue borders are previously unreported mutations.
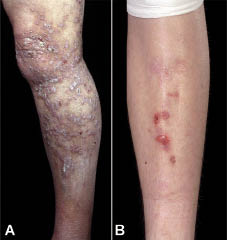
Fig. 3. Phenotypic heterogeneity for the COL7A1 glycine substitution mutation p.G2043R. (A) In this individual heterozygosity for the p.G2043R mutation results in epidermolysis bullosa pruriginosa (EBP) with lichenified prurigo-like lesions on the leg. (B) In contrast, the same mutation in another individual results in non-EBP dominant dystrophic epidermolysis bullosa (DEB), here showing minor blistering and scarring on the shin.
Table II. Incidence of the 1G/2G polymorphism at the –1607 position of the MMP1 gene promoter in non-EBP DDEB, dominant EBP, RDEB and controls in this study
|
|
EBP
|
Non EBP-DDEB
|
RDEB
|
Controls
|
|
Total patient no.
|
27
|
23
|
25
|
50
|
|
Total allele no.
|
54
|
46
|
50
|
100
|
|
1G/1G
|
7 (25.9%)
|
6 (26.1%)
|
2 (8.0%)
|
16 (32.0%)
|
|
1G/2G
|
15 (55.6%)
|
10 (43.5%)
|
15 (60.0%)
|
26 (52.0%)
|
|
2G/2G
|
5 (18.5%)
|
7 (30.4%)
|
8 (32.0%)
|
8 (16.0%)
|
|
Total 2G alleles/
Total 1G+2G alleles
|
25/54
|
24/46
|
31/50
|
42/100
|
|
GG allelic frequency (%)
|
46.3
|
52.2
|
62*
|
42
|
*p = 0.02 compared with controls.
DDEB: dominant dystrophic epidermolysis bullosa; RDEB: recessive dystrophic epidermolysis bullosa; EBP: epidermolysis bullosa pruriginosa.
Genotyping of the MMP1 polymorphism in the control group showed that the major allele was 1G (58.0%) and the minor allele was 2G (42.0%). In the EBP group, sequencing showed that the frequency of 1G was 53.7% and of 2G was 46.3%, whereas in the non-EBP dominant DEB individuals the frequency of 1G was 47.8% and of 2G was 52.2%. For the recessive DEB group the allele frequencies were 38.0% for 1G and 62.0% for 2G. These data are shown in more detail in Table II. No significant difference in allelic frequency was found between EBP and controls (p = 0.61), between EBP and non-EBP dominant DEB (p = 0.56), or between non-EBP dominant DEB and controls (p = 0.25). However, a significant difference in the incidence of allelic frequency was observed between the recessive DEB group and controls (p = 0.02): the frequency of the 2G allele is clearly higher in the recessive DEB subjects.
With respect to the MMP1 genotype in individuals with the COL7A1 mutation p.G2043R, 10 out of 12 individuals expressed the 2G variant on at least one allele but the two subjects who were homozygous for 1G had non-EBP dominant DEB. Differences between the 2 phenotypes with respect to the 1G/2G alleles were not significant. Inter-familial phenotypic heterogeneity was also observed for the COL7A1 mutation p.G1522E. Individuals who were heterozygous for this glycine substitution had either EBP or bullous dermolysis of the newborn (OMIM 131705), but this considerable phenotypic disparity was not explained by the MMP1 polymorphism as the 2G allele was detected in both clinical variants. Intra-familial variability in phenotype was noted in one family with EBP that was heterozygous for the COL7A1 mutation p.G2251E. Clinicopathological details of this family have been reported previously (11). However, there was no correlation between the presence or absence of the 2G allele and the EBP phenotype.
DISCUSSION
This study has identified six new heterozygous glycine substitutions in type VII collagen as the molecular basis for EBP. It also identifies another 6 novel heterozygous glycine substitutions underlying non-pruritic dominant DEB (Table I). These findings expand the total COL7A1 glycine substitution mutation database to 133 mutations (71 dominant, 62 recessive) (29–31), but demonstrate that there is nothing atypical in the nature or location of the EBP mutations compared with other dominant or recessive forms of DEB resulting from glycine substitution mutations in type VII collagen (29–33).
In addition, our data on the 1G/2G MMP1 gene promoter polymorphism show that there is no association between the 2G allele and the EBP phenotype. Thus, the striking inflammatory and itchy skin phenotype of EBP does not appear to be explained by this potential genetic modifier.
Nevertheless, MMPs may contribute to various pathophysiological processes in DEB, such as cancer progression (squamous cell carcinoma) and wound healing (34–37). For example, MMP7 has the capacity to cleave surface molecules such as E-cadherin and syndecan-1, as well as extracellular matrix proteins including fibrillin and type VII collagen, thereby facilitating cancer progression (34, 35). Furthermore, varying clinical severity in three siblings affected with recessive DEB has been shown to correlate with increased levels of MMP2, MMP3 and MMP9 and reduced levels of the tissue inhibitor of metalloproteinase-1 (TIMP1) (36). In contrast, however, our study has demonstrated no correlation between specific clinical phenotypes of dominant DEB and a functional polymorphism in the MMP1 gene promoter. The possibility of genetic variants in these other MMPs contributing to phenotypic expression awaits further study.
EBP can be a difficult disorder to treat. Previous treatments have included topical and systemic corticosteroids, topical tacrolimus, ciclosporin and thalidomide. In our 27 subjects with EBP, treatment with topical tacrolimus in eight individuals provided significant symptomatic relief from the pruritus in only one subject, and partial relief in three others. Similar responses to topical steroids were also noted. Systemic therapies were tried in too few of these particular individuals to comment on their efficacy.
Based on the data from Titeux et al. (18), novel anti-proteolytic therapies may emerge as useful treatment options for some individuals with DEB, especially perhaps for recessive DEB, but our findings (albeit based on findings in genomic DNA) do not suggest that EBP subjects will specifically benefit from this form of treatment compared with other individuals with DEB.
Our findings in recessive DEB, however, add support to the published data showing that the 2G polymorphism may be associated with greater clinical severity (18). But there may also be other clinical implications, perhaps relevant to the pathophysiology of squamous cell carcinoma, which is a frequent complication in this subtype of DEB (37). Within the recessive DEB group that we assessed, six subjects had developed cutaneous squamous cell carcinoma before the age of 30 years. Of note, each of these individuals had the 2G variant on one or both MMP1 alleles. This observation is of interest, given the potential role of MMPs in cancer invasion through degradation of the extracellular matrix (20–23). Indeed, in other studies, the 2G polymorphism has been associated with an increased risk of developing colorectal, oesophageal, bladder, breast, lung and head and neck cancers (38–42). Furthermore, for some of these malignancies the expression level of MMP1 has been found to correlate negatively with survival in affected patients (43–46). These findings led to the possibility of trying to modify cancer progression by using pharmacological inhibitors of MMPs. Despite promising data in animal models, however, the lack of specificity of the currently available inhibitors, and the adverse effects noted in clinical trials, have so far failed to yield significant benefits for patients (47). Nevertheless, specific anti-cancer agents targeting MMPs, TIMPs or genetic regulators of MMPs such as the tumour suppressor gene RECK, may have future relevance to the management of squamous cell carcinoma in recessive DEB.
With regards to the EBP phenotype, however, our study has shown that the unusual inflammatory clinical features seen in this condition are not explained by a common genetic variant in the MMP1 gene promoter and the pathophysiology of this subtype of DEB therefore remains unresolved.
ACKNOWLEDGEMENTS
We are grateful to all individuals who participated in this study. Dr Noor Almaani is a clinical research fellow supported by the Dystrophic Epidermolysis Bullosa Research Association (DebRA, UK).
REFERENCES
1. Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): report of the third international consensus meeting on diagnosis and classification of EB. J Am Acad Dermatol 2008; 58: 931–950.
2. Uitto J, Pulkkinen L, Christiano AM. Molecular basis of the dystrophic and junctional forms of epidermolysis bullosa: mutations in the type VII collagen and kalinin (laminin 5) genes. J Invest Dermatol 1994; 103: 39S–46S.
3. Christiano AM, Ryynanen M, Uitto J. Dominant dystrophic epidermolysis bullosa: Identification of a Gly–Ser substitution in the triple-helical domain of type VII collagen. Proc Natl Acad Sci 1994; 91: 3549–3553.
4. McGrath JA, Schofield OM, Eady RA. Epidermolysis bullosa pruriginosa: dystrophic epidermolysis bullosa with distinctive clinicopathological features. Br J Dermatol 1994; 130: 617–625.
5. Lee JY, Pulkkinen L, Liu HS, Chen YF, Uitto J. A glycine-to-arginine substitution in the triple-helical domain of type VII collagen in a family with dominant dystrophic epidermolysis bullosa pruriginosa. J Invest Dermatol 1997; 108: 947–949.
6. Mellerio JE, Ashton GH, Mohammedi R, Lyon CC, Kirby B, Harman KE, et al. Allelic heterogeneity of dominant and recessive COL7A1 mutations underlying epidermolysis bullosa pruriginosa. J Invest Dermatol 1999; 112: 984–987.
7. Drera B, Castiglia D, Zoppi N, Gardella R, Tadini G, Floriddia G, et al. Dystrophic epidermolysis bullosa pruriginosa in Italy: clinical and molecular characterization. Clin Genet 2006; 70: 339–347.
8. Schumann H, Has C, Kohlhase J, Bruckner-Tuderman L. Dystrophic epidermolysis bullosa pruriginosa is not associated with frequent FLG gene mutations. Br J Dermatol 2008; 159: 464–469.
9. Chuang GS, Martinez-Mir A, Yu HS, Has C. A novel missense mutation in the COL7A1 gene underlies epidermolysis bullosa pruriginosa. Clin Exp Dermatol 2004; 29: 304–307.
10. Dang N, Klingberg S, Marr P, Murrell DF. Review of collagen VII sequence variants found in Australasian patients with dystrophic epidermolysis bullosa reveals nine novel COL7A1 variants. J Dermatol Sci 2007; 46: 169–178.
11. Ee HL, Liu L, Goh CL, McGrath JA. Clinical and molecular dilemmas in the diagnosis of familial epidermolysis bullosa pruriginosa. J Am Acad Dermatol 2007; 56: S77–S81.
12. Jiang W, Bu D, Yang Y, Zhu X. A novel splice site mutation in collagen type VII gene in a Chinese family with dominant dystrophic epidermolysis bullosa pruriginosa. Acta Derm Venereol 2002; 82: 187–191.
13. Murata T, Masunaga T, Shimizu H, Takizawa Y, Ishiko A, Hatta N, et al. Glycine substitution mutations by different amino acids in the same codon of COL7A1 lead to heterogeneous clinical phenotypes of dominant dystrophic epidermolysis bullosa. Arch Dermatol Res 2000; 292: 477–481.
14. Nakamura H, Sawamura D, Goto M, Sato-Matsumura KC, LaDuca J, Lee JY, et al. The G2028R glycine substitution mutation in COL7A1 leads to marked inter-familiar clinical heterogeneity in dominant dystrophic epidermolysis bullosa. J Dermatol Sci 2004; 34: 195–200.
15. Whittock NV, Ashton GH, Mohammedi R, Mellerio JE, Mathew CG, Abbs SJ, et al. Comparative mutation detection screening of the type VII collagen gene (COL7A1) using the protein truncation test, fluorescent chemical cleavage of mismatch, and conformation sensitive gel electrophoresis. J Invest Dermatol 1999: 113: 673–686.
16. Ren X, Liu JY, Zhai LY, Yao Q, Dai X, Cai Z, et al. A splicing mutation in the COL7A1 gene causes autosomal dominant dystrophic epidermolysis bullosa pruriginosa. Br J Dermatol 2008; 158: 618–620.
17. Lapinski P, Lapiere JC, Traczyk T, Chan LS. Sporadic dystrophic epidermolysis bullosa with concomitant atopic dermatitis. Br J Dermatol 1998; 138: 315–320.
18. Titeux M, Pendaries V, Tonasso L, Décha A, Bodemer C, Hovnanian A. A frequent functional SNP in the MMP1 promoter is associated with higher disease severity in recessive dystrophic epidermolysis bullosa. Hum Mutat 2008; 29: 267–276.
19. Seltzer JL, Eisen AZ, Bauer EA, Morris NP, Glanville RW, Burgeson RE. Cleavage of type VII collagen by interstitial collagenase and type IV collagenase (gelatinase) derived from human skin. J Biol Chem 1989; 264: 3822–3826
20. Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T. Regulation of matrix metalloproteinases: an overview. Mol Cell Biochem 2003; 253: 269–285.
21. Ghilardi G, Biondi ML, Mangoni J, Leviti S, DeMonti M, Guagnellini E, et al. Matrix metalloproteinase-1 promoter polymorphism 1G/2G is correlated with colorectal cancer invasiveness. Clin Cancer Res 2001; 7: 2344–2346.
22. Ye S, Dhillon S, Turner SJ, Bateman AC, Theaker JM, Pickering RM, et al. Invasiveness of cutaneous malignant melanoma is influenced by matrix metalloproteinase 1 gene polymorphism. Cancer Res 2001; 61: 1296–1298.
23. Zinzindohoué F, Lecomte T, Ferraz JM, Houllier AM, Cugnenc PH, Berger A, et al. Prognostic significance of MMP-1 and MMP-3 functional promoter polymorphisms in colorectal cancer. Clin Cancer Res 2005; 15: 594–599.
24. Sambrook J, Fritsch EF, Maniatis T, editors. Molecular cloning. A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 1989.
25. Christiano AM, Hoffman GG, Zhang X, Xu Y, Tamai Y, Greenspan DS, et al. Strategy for identification of sequence variants in COL7A1 and a novel 2-bp deletion mutation in recessive dystrophic epidermolysis bullosa. Hum Mutat 1997; 10: 408–414.
26. Rutter JL, Mitchell TI, Butticè G, Meyers J, Gusella JF, Ozelius LJ, et al. A single nucleotide polymorphism in the matrix metalloproteinase-1 promoter creates an Ets binding site and augments transcription. Cancer Res 1998; 58: 5321–5325.
27. Mellerio JE, Salas-Alanis JC, Talamantes ML, Horn H, Tidman MJ, Ashton GH, et al. A recurrent glycine substitution mutation, G2043R, in the type VII collagen gene (COL7A1) in dominant dystrophic epidermolysis bullosa. Br J Dermatol 1998; 139: 730–737.
28. Murata T, Masunaga T, Ishiko A, Shimizu H, Nishikawa T. Differences in recurrent COL7A1 mutations in dystrophic epidermolysis bullosa: ethnic-specific and worldwide recurrent mutations. Arch Dermatol Res 2004; 295: 442–447.
29. Dang N, Murrell DF. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp Dermatol 2008; 17: 553–568.
30. Whittock NV, Ashton GH, Mohammedi R, Mellerio JE, Mathew CG, Abbs SJ, et al. Comparative mutation detection screening of the type VII collagen gene (COL7A1) using the protein truncation test, fluorescent chemical cleavage of mismatch, and conformation sensitive gel electrophoresis. J Invest Dermatol 1999; 113: 673–686.
31. Christiano AM, McGrath JA, Tan KC, Uitto J. Glycine substitutions in the triple-helical region of type VII collagen result in a spectrum of dystrophic epidermolysis bullosa phenotypes and patterns of inheritance. Am J Hum Genet 1996; 58: 671–681.
32. Varki R, Sadowski S, Uitto J, Pfendner E. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype-genotype correlations in the dystrophic subtypes. J Med Genet 2007; 44: 181–192.
33. Uitto J, Richard G. Progress in epidermolysis bullosa: genetic classification and clinical implications. Am J Med Genet C Semin Med Genet 2004; 131C: 61–74.
34. Kivisaari AK, Kallajoki M, Mirtti T, McGrath JA, Bauer JW, Weber F, et al. Transformation-specific matrix metalloproteinases (MMP)-7 and MMP-13 are expressed by tumour cells in epidermolysis bullosa-associated squamous cell carcinomas. Br J Dermatol 2008; 158: 778–785.
35. Changotade SI, Assoumou A, Guéniche F, Fioretti F, Séguier S, de Prost Y, et al. Epigallocatechin gallate’s protective effect against MMP7 in recessive dystrophic epidermolysis bullosa patients. J Invest Dermatol 2007; 127: 821–828.
36. Bodemer C, Tchen SI, Ghomrasseni S, Séguier S, Gaultier F, Fraitag S, et al. Skin expression of metalloproteinases and tissue inhibitor of metalloproteinases in sibling patients with recessive dystrophic epidermolysis and intrafamilial phenotypic variation. J Invest Dermatol 2003; 121: 273–279.
37. Mallipeddi R. Epidermolysis bullosa and cancer. Clin Exp Dermatol 2002; 27: 616–623.
38. Hinoda Y, Okayama N, Takano N, Fujimura K, Suehiro Y, Hamanaka Y, et al. Association of functional polymorphisms of matrix metalloproteinase (MMP)-1 and MMP-3 genes with colorectal cancer. Int J Cancer 2002; 102: 526–529.
39. Hashimoto T, Uchida K, Okayama N, Imate Y, Suehiro Y, Hamanaka Y, et al. Association of matrix metalloproteinase (MMP)-1 promoter polymorphism with head and neck squamous cell carcinoma. Cancer Lett 2004; 211: 19–24.
40. Przybylowska K, Kluczna A, Zadrozny M, Krawczyk T, Kulig A, Rykala J, et al. Polymorphisms of the promoter regions of matrix metalloproteinases genes MMP-1 and MMP-9 in breast cancer. Breast Cancer Res Treat 2006; 95: 65–72.
41. Tasci AI, Tugcu V, Ozbek E, Ozbay B, Simsek A, Koksal V. A single-nucleotide polymorphism in the matrix metalloproteinase-1 promoter enhances bladder cancer susceptibility. BJU Int 2008; 101: 503–507.
42. Zhu Y, Spitz MR, Lei L, Mills GB, Wu X. A single nucleotide polymorphism in the matrix metalloproteinase-1 promoter enhances lung cancer susceptibility. Cancer Res 2001; 61: 7825–7829.
43. Murray GI, Duncan ME, O’Neil P, McKay JA, Melvin WT, Fothergill JE. Matrix metalloproteinase-1 is associated with poor prognosis in oesophageal cancer. J Pathol 1998; 185: 256–261.
44. Murray GI, Duncan ME, O’Neil P, Melvin WT, Fothergill JE. Matrix metalloproteinase-1 is associated with poor prognosis in colorectal cancer. Nat Med 1996; 2: 461–462.
45. Kanamori Y, Matsushima M, Minaguchi T, Kobayashi K, Sagae S, Kudo R, et al. Correlation between expression of the matrix metalloproteinase-1 gene in ovarian cancers and an insertion/deletion polymorphism in its promoter region. Cancer Res 1999; 59: 4225–4227.
46. Cheng S, Tada M, Hida Y, Asano T, Kuramae T, Takemoto N, et al. High MMP-1 mRNA expression is a risk factor for disease-free and overall survivals in patients with invasive breast carcinoma. J Surg Res 2008; 146: 104–109.
47. Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 2002; 295: 2387–2392.